Current Issue : Article / Volume 3, Issue 1
- Research Article | DOI:
- https://doi.org/10.58489/2836-5038/013
Characterisation of Stromal Cells Which Support Hematopoietic Niches in Murine Spleen
1Clem Jones Centre for Regenerative Medicine, Bond University, Gold Coast, QLD, Australia.
2Research School of Biology, Australian National University, Canberra ACT, Australia.
Helen C OâNeill*
Hong Kiat Lim, Helen C OâNeill. (2024). Characterisation of Stromal Cells Which Support Hematopoietic Niches in Murine Spleen. International Journal of Stem Cells and Medicine. 3(1); DOI: 10.58489/2836-5038/013
© 2024 Helen C OâNeill, this is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
- Received Date: 11-07-2024
- Accepted Date: 19-07-2024
- Published Date: 29-07-2024
Spleen, hematopoiesis, stromal cells, hematopoietic stem cell, niches
Abstract
The role of spleen in hematopoiesis is not well understood. Recent studies now identify the presence of hematopoietic stem cells (HSC) in spleen although their numbers are far lower than HSC in bone marrow. Neonatal and pregnant mice contain higher numbers of HSC than do normal adults and have been used here as a model to investigate the localisation of HSC within the spleen.
Immunocytochemical staining of spleen sections has been used to identify stromal cell types in the microenvironment of spleen. These studies have demonstrated the stromal structure and splenic architecture of pregnant adult mice and neonatal mice compared with normal adult mouse spleen. Six-day (D6) neonatal spleen is characterised by small T and B cell areas in poorly formed white pulp areas, and a surfeit of red pulp marked by the presence of numerous sinusoids and stromal cells, and notably the presence of gp38-staining cells. These are absent from adult red pulp region, being confined to the white pulp region as fibroblastic reticular cells. Another model tested was extramedullary hematopoiesis induced in adult mouse given Flt3L. Quantification of hematopoietic stem and progenitor cells (HSPC) in the various models revealed that pregnant mice contained significantly more long-term (LT)-HSC than did neonatal or adult normal mice, and that the spleen of Flt3L treated mice was populated more by multipotential progenitors (MPP) than LT-HSC.
The pregnant and neonatal mice were subsequently analysed for localisation of LT-HSC in relation to various stromal cell types including endothelial cells (CD31), angiogenic vasculature (CD105), mesenchymal cells (CD29), fibroblastic reticular cells (gp38), myeloid cells (F4/80) and perivascular reticular cells (CD140b, CD51, Thy1.2/CD90). Multi-colour staining was used to colocalise LT-HSC with different region-specific stromal subsets. In both neonatal and pregnant mice, LT-HSC were located in red pulp. While many HSC colocalised with CD140b-exppressing stroma, some were found in association with myeloid cells, Thy1.2+ cells and in the case of neonates, with gp38+ cells.
CD140-expressing cells in spleen were investigated more fully through gene expression analysis using the Fluidigm platform. Clustering analysis of expressed genes identified 3 distinct cell types, one reflecting follicular dendritic cells that do not support HSC niches. These were in the white pulp of adult mice. Two further clusters reflected perivascular reticular cells, one subset expressing type-specific genes as well as mesenchymal lineage genes at a higher level. It was concluded that a subset of mature and immature perivascular reticular cells cells may exist in spleen. These data confirm the presence of HSC and HSC niches in spleen and identify mesenchymal CD140b-expressing perivascular reticular cells as important niche elements in line with similar findings on HSC niches in bone marrow.
Introduction
Stem cell niches refer to a microenvironment comprising non-hematopoietic cells which interact with hematopoietic stem cells (HSC) via juxtacrine and paracrine signalling to regulate self-renewal and differentiation, as well as proliferation and quiescence [1]. In the bone marrow, hematopoietic niches reflect endosteal, perisinusoidal and periarteriolar microenvironments, and involve cells like osteoblasts, endothelial cells and perivascular reticular cells [2-8]. In bone marrow, the perisinusoidal niche maintains HSC and other hematopoietic progenitors, and the stromal components have been described as nestin+ mesenchymal stem cells, leptin receptor+ stromal cells and CXCL12-abundant reticular (CAR) cells [3-5, 8, 9]. These cell types have been shown by immunofluorescence imaging to lie in close proximity to HSC [3-5], and to be localised adjacent to arterioles and sinusoids [3, 4, 9]. They are now generally classified as perivascular reticular cells.
While HSC niches in bone marrow have been extensively studied, limited knowledge is available on niches for HSC in spleen. HSC have been localised in close contact with sinusoids in the red pulp of spleen [8, 10], and a perisinusoidal niche for HSC has been associated with extramedullary hematopoiesis [10]. Spleen contains HSC niches, similar to those of bone marrow comprising both mesenchymal perivascular cells and the endothelial cells of sinusoids [11]. Previous studies in this laboratory have determined that a stromal cell derivative of a murine, splenic long-term culture, termed 5G3, which resembles perivascular reticular cells, can support restricted in vitro hematopoiesis and production of specific myeloid cell subsets [12-14]. In addition, stromal fractions isolated from neonatal spleen were also shown to support in vitro hematopoiesis when overlaid with lineage-negative bone marrow cells, albeit at lower levels than achieved with the 5G3 stromal cell line (HL). The gene profile and phenotype of 5G3 stroma reflects a mesenchymal cell expressing genes common to perivascular reticular cells like Cxcl12, Scf, Pdgfrb, Pdgfra, Endoglin, Itagv, Sca-1 and Thy1.2 [15], so that it may overlap with Tcf21+Pdgfrαβ+ cells, described by Inra et al (2015) as mesenchymal cells which form the perisinusoidal niche in the red pulp region of spleen [10].
Extramedullary hematopoiesis occurs as a natural process during fetal development and involves yolk sac, fetal liver and spleen [16]. Extramedullary hematopoiesis is also activated during stress conditions including infection and pregnancy, and is evidenced by studies showing that the low number of HSPC present in murine spleen in the steady-state increases quickly following inflammation [17, 18]. The presence of HSPC in spleen in the steady-state is not restricted to mouse since spleens of pigs, baboons and humans were also found to retain a low number of HSPC under steady-state conditions [19]. Moreover, in cell tracing experiments, spleen cells derived from both neonatal and adult mice were able to provide hematopoietic reconstitution of lethally irradiated host mice following adoptive transfer [20, 21], confirming that spleen can house HSPC in the resting state, and can also increase numbers of HSPC during times of stress or inflammation. Evidence suggesting a role for spleen in hematopoiesis raises the possibility that splenic HSC niches can support maintenance of HSC both in the resting state, and following influx when extramedullary hematopoiesis is activated. Characteristics of splenic stroma in mice under these conditions has been investigated here to determine the defining characteristics of hematopoietic niches. Here, stromal cell niches for HSC have been investigated in neonatal and adult C57BL/6J mice, in pregnant mice undergoing extramedullary hematopoiesis, and in mice in which bone marrow HSPC have been mobilised out of bone marrow through administration of Fms-like tyrosine kinase (Flt)-3 ligand (Flt3L). The increase in oestrogen during pregnancy induces extramedullary hematopoiesis marked by increased numbers of HSC, higher HSC cell division and erythropoiesis [22]. Flt3L induction of HSPC mobilisation reflects a model for extramedullary hematopoiesis in which higher numbers of HSC are expected to localise in spleen [23-25].
Immunofluorescence imaging has been used here to localise HSC, and to determine their spatial relationship with known stromal cell types in the spleen. Spleen sections were stained with CD29, Sca-1(Ly6A) and CD54/ICAM-1, known markers of many mesenchymal cell subsets in spleen [26, 27], CD105 which marks angiogenic vasculature [28], CD31 which identifies mature endothelial cells in both the red and white pulp regions of spleen [26, 29], gp38 (podoplanin) which identifies fibroblastic reticular cells [26, 30], CD140a/b (PDGFRa/b) which identifies fibroblastic reticular cells, red pulp fibroblasts, perivascular reticular cells and mesenchymal stem cells [10, 26, 27, 31], as well as CD90/Thy-1 which identifies mesenchymal stem cells, as well as T cells [31-34]. Immunofluorescence imaging has been used to localise primitive long-term (LT)-HSC in spleen through their well described Lineage-c-Kit+Sca-1+CD41/48-CD150+ phenotype [8, 35, 36]. By using a combination of markers for stromal and hematopoietic cells it has been possible to identify differences in the stromal architecture of neonatal, adult and pregnant mice. LT-HSC have also been shown to be present in highest number in pregnant mice. The CD140a/b (PDGFRa/b)+ stromal cells which identify niches for HSC were further investigated through gene expression analysis to identify several subset types amongst these cells, raising the possibility of distinct niches within spleen.
Materials and Methods
Animals and spleen cell isolation
Specific pathogen-free C57BL/6J (H-2Kb) mice aged 6-weeks, or 6-days (D6), were obtained from the John Curtin School of Medical Research (JCSMR: Canberra, Australia). Pregnant mice were used at embryonic day (E)18. Fms-like tyrosine kinase (Flt)-3 ligand treatment involved giving mice a subcutaneous inoculum of B16-Ft3L melanoma cells which secrete Flt3L [37]. After 7 days mice were sacrificed for analysis of HSC present in spleen. This treatment is known to mobilise HSC into blood and spleen [23, 25]. All mice were housed and handled according to Protocol #A2013/11 approved by the Animal Experimentation Ethics Committee at the Australian National University (ANU: Canberra, Australia). For isolation of splenocytes, dissected spleen was pressed through a fine mesh sieve. Cells were resuspended into 1mL medium and treated with RBC lysis buffer, washed and counted. Cells from at least 6 animals were pooled for each splenocyte preparation.
Stromal cell isolation
Murine spleens were dissociated between two sterile microscopic slides before filtering through a 70µm cell strainer. The stromal fraction was collected in a tube containing 2mL collagenase IV extraction buffer [(2% heat inactivated fetal calf serum, 1 mg/mL collagenase IV (Sigma-Aldrich: Castle Hill, NSW, Australia) and 40µg/mL DNaseI (Sigma-Aldrich) in RPMI medium (REF), and incubated for 20 minutes at 37oC with rotation. 2mL collagenase D extraction buffer [2% heat inactivated fetal calf serum, 1 mg/mL collagenase D (Roche Applied Science: North Ryde, NSW, Australia) and 40µg/mL DNaseI in RPMI medium] was added for another 20-minute incubation at 37oC with rotation. An additional 2mL of collagenase D extraction buffer was added and the cell suspension incubated for a further 20 minutes at 37oC with rotation. The activity of collagenase was halted by addition of 60µl of 500mM EDTA to give a final concentration of 5mM. Dissociated stromal cells were washed twice with 5mL sDMEM [Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 4g/L D-glucose, 6mg/L folic acid, 36mg/L L-asparagine, 116mg/L L-arganine HCL, 10% heat inactivated fetal calf serum (JRH Biosciences: Lenexa, Kansas, USA), 10mM Hepes, 2mM L-glutamine, 100 U/mL penicillin, 100µg/mL streptomycin and 5x10-5M 2-mercaptoethanol]. Washing involved centrifugation (300g, 4oC, 5 minutes). Viable cell count was determined using trypan blue staining and the cell pellet resuspended in 1mL sDMEM for antibody staining, flow cytometric analysis and cell sorting.
Antibodies
Antibodies used for cell staining were purchased as affinity purified conjugates of either fluorochrome or biotin, along with isotype control antibodies and secondary fluoresceinated conjugates from Biolegend (San Diego, CA, USA). All antibodies were titrated before use on splenocytes to determine the concentration required to give minimum saturation binding. Antibodies used for flow cytometry had specificity for CD45.2 (clone 104), c-Kit (2B8), Sca-1 (Ly6A: D7), CD150 (TC15-12F12.2), CD41 (MWReg30), CD48 (HM48-1), Flt3(A2F10), CD140a (APA5) and CD140b (APB5). Antibodies used for immunocytochemistry were specific for CD3e (145-2C11), B220 (RA3-6B2), F4/80 (BM8), CD29 (HMb1-1), c-Kit(2B8), CD150 (TC15-12F12.2), CD41 (MWReg30), CD48 (HM48-1), CD31 (390), gp38 (8.1.1), CD105 (MJ7/18), Thy1.2 (30-H12), CD140b (APB5) and Sca-1 (Ly6A: D7). Lineage (Lin) staining of cells involved the Lineage Cell Depletion kit (Miltenyi Biotec, Bergisch-Gladbach, Germany). This cocktail of biotinylated antibodies was used to stain hematopoietic cells expressing CD5, B220, CD11b, Gr-1, Ly6C/G, 7-4 and Ter119 for magnetic bead depletion. The procedure followed supplier instructions. Magnetic bead depleted cells were subsequently sorted using gating on the flow cytometer for further purification of the Lineage-depleted population. The Lineage Cell Depletion kit (Miltenyi Biotec) followed by a secondary labelled conjugate was also used to stain sections for immunocytochemistry.
Flow cytometric analysis of cell surface marker expression
For staining, 5x105 - 1x106 cells in FACS buffer [1 percent fetal calf serum, 0.1% sodium azide in sDMEM] were sedimented into the wells of a 96-well polystyrene microtitre plate (Corning: New York, USA) by centrifugation (1300 rpm, 4 oC, 5 minutes). Supernatant was discarded and cells resuspended in 25µl anti-CD16/32 (FcBlock) (Biolegend) at 5µg/mL with incubation on ice for 15 minutes. The plate was centrifuged (1300 rpm, 4oC, 5 minutes), and supernatant discarded. Cells were then resuspended in 25µl of diluted primary antibody and incubated on ice for 25 minutes. Cells were washed twice with 150µl/106 cells FACS buffer with centrifugation (1300rpm, 4oC, 5 minutes). Where necessary, 25µl of diluted secondary antibody or conjugate was added to each well, and cells incubated on ice for 25 minutes. Cells were then washed again using the washing procedure and resuspended in 50µl FACS buffer with transfer to FACS cluster tubes (Corning) for analysis. For multicolour staining, up to five different primary antibodies were used. Where necessary, propidium iodide (PI: 1µg/mL) was added for dead cell discrimination on the flow cytometer.
Cells labelled with fluorochrome-labelled antibodies specific for markers of interest were analysed using an LSRII flow cytometer (Becton Dickinson: Franklin Lakes, New Jersey, USA). Parameters of voltage and event counts were preset using FACSDIVA software (Becton Dickson). For multicolour analysis, compensation was carried out manually using single colour controls. Isotype control antibodies were used to determine the background binding of each specific antibody and to set gates for delineation of positively stained cells. Live and dead cells were discriminated by exclusion of propidium iodide (PI). Analysis of data was performed using FACSDiva and Flow Jo software (Tristar: Phoenix, Arizona, USA). Data was collected as bivariate plots of side scatter (SSC), fluorescence or forward scatter (FSC). Percent positive cells was calculated using Flow Jo software (Tristar).
Immunofluorescence microscopy
Whole C57BL/6J adult or neonatal murine spleen was embedded in Tissue-Tek OCTTM compound [10.24% polyvinyl alcohol, 4.26% polyethylene glycol, 85.50% non-reactive ingredient]. Sections of 10µm thickness were then prepared using a Frigocut 2800 cryostat (Reichert-Jung: Depew, New York, USA). Spleen sections were then acetone-fixed for 15 minutes at 4oC and air dried for an hour before storage at -80oC. Frozen slides were left to thaw at room temperature before staining for immunofluorescence microscopy.
For antibody staining, tissue sections were first blocked with 10% heat inactivated fetal calf serum in PBS for 45 minutes at room temperature to prevent non-specific binding of antibodies. Sections were then stained with fluorochrome-labelled primary antibodies specific to antigens of interest for 45 minutes at room temperature. Slides were then washed thrice by immersion in three consecutive chambers containing PBS, for 5 minutes each. Secondary antibodies were used to stain sections where necessary (for 45 minutes) and washed again as above. Nuclear DNA was stained with 4, 6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) for 5 minutes and washed thrice with PBS as above to delineate individual cell nuclei. Stained sections were examined using a Leica TCS SP5 Confocal microscope (Leica Microsystems: North Ryde, NSW, Australia).
Single-cell transcriptome analysis
Collagenase isolated splenic stromal cells were prepared for antibody staining. Cells were stained with the Lineage Depletion cocktail and enriched by magnetic bead depletion. The enriched cell population was then stained with Lineage antibodies as well as fluoresceinated antibodies to markers of interest. After washing, cells were stained with propidium iodide (PI) to identify dead cells and then run through a BD FACSAria™ II cell sorter (Becton Dickinson). Sorting involved initial gating out of dead (PI-) cells and doublets (FSC-A and FSC-H gating). Live cells were identified by marker expression, and gates for sorting were set using isotype control antibodies.
Sorted stromal cells were suspended in 1mL sDMEM, the cells centrifuged and resuspended in 10μL of sDMEM for loading into the Fluidigm C1 Single-Cell Auto Prep IFC chip (5-10μm cell diameter) (Millennium Science: Victoria, Australia). The loaded chip was imaged with a phase-contrast microscope to confirm cell integrity and capture. cDNA was prepared from each cell on the chip using the SMARTer Ultra Low RNA kit for Fluidigm C1 system (Scientifix Life, Victoria, Australia). This step was carried out automatically in the Fluidigm C1 system. The mRNA library was constructed and sequenced using high output Next Seq 500 with a read length of 75 bp, and 10 million reads per sample (Biomolecular Resources Unit: JCSMR, Canberra, ACT, Australia).
Heat map analysis and agglomerative hierarchical clustering was performed using Ward’s minimum variance method and involved the use of R (http://www/r-project.org). Data mining on the basis of signal values was used to determine gene expression associated with known functions of cell lineages.
Statistical analysis
Data are presented as mean + standard error. The Student t-test was used to assess significance (p less then 0.05) using Prism 5.0 software (Graphpad Software: San Diego, CA, USA).
Results
Stromal cell organisation in neonatal compared with adult spleen
Initially single colour staining of spleen sections was used to optimise antibodies and to test their capacity to discriminate spleen architecture (Figure S1). These studies revealed distinct stromal cell organisation in neonatal spleen and associated distribution of hematopoietic cells typical of developing neonates. Subsequent studies then used multi-colour staining to verify differences in splenic architecture between different aged groups of mice.
The adult spleen is uniquely arranged in a way that reflects its functional capacity in hematopoiesis and immunity. Three main regions can be delineated including B cell follicles, T cell zones and red-pulp regions, using antibodies specific for the B220, CD3 and F4/80 markers, respectively (Figure 1a). Similar staining of 6-day old neonatal spleen indicated more extensive red pulp areas containing fewer F4/80+ myeloid cells, with smaller follicles showing separated B cell follicles and T cell zones (Figure 1b). Red pulp in adult spleen was marked by an extensive network of Ly6A/Sca-1 and CD29 staining stromal cells in close proximity to F4/80+ myeloid cells (Figure 1c, c’, c’’). In contrast, the more extensive red pulp region of neonatal spleen was dominated by myeloid cells with very few mesenchymal stromal cells indicated by weak staining with CD29 and Ly6A/Sca-1 (Figure 1d, d’, d’’). Adult spleen was also characterised by the presence of scattered CD29+ fibroblastic reticular cells in the white pulp, and densely staining CD29+Ly6A/Sca-1+ marginal zones (Figure 1c’’). By contrast, in neonatal spleen, CD29+Ly6A/Sca-1+ staining cells colocalised with the white pulp region and the central arteriole (Figure 1d, d’, d’’). CD29+Sca-1+ staining cells could be detected in red pulp although these were rare (Figure 1d’’’). While neonatal spleen reveals white pulp formation, the stromal network is distinctly different from adult spleen, both in terms of cell distribution and cell phenotype. This likely represents a developmental stage in formation of the distinct stromal subsets which underlie the spleen and support its role in hematopoiesis and immune response development.
HSC in spleen
The red pulp region of spleen is known to harbour HSC [10]. Since the red pulp was more extensive in neonatal over adult mice, we questioned whether neonates might house more HSC and so prove a better model for the study of hematopoietic niches. To this end, we also studied two models of extramedullary hematopoiesis, namely pregnant mice and mice given Flt3L to mobilise HSPC into blood and spleen. These two models have been reported to have higher numbers of HSC in spleen [22-25].
While Ly6A/Sca-1 and c-Kit along with lineage markers, have been routinely used in flow cytometry to delineate bone marrow HSC, a report by Morita and colleagues [38] has confirmed that bone marrow HSC are phenotypically similar to splenic HSC. Therefore, antibodies to Sca-1 and c-Kit were used initially to delineate splenic HSC in spleen sections, along with the F4/80 marker of myeloid cells to delineate the red pulp region. Adult spleen sections stained with antibody for Ly6A/Sca-1, c-Kit, and F4/80 showed no staining for Sca-1+c-Kit+ cells reflecting HSC, with Sca-1+ stromal cells present in the red pulp, and Sca-1 strongly staining the marginal zone region of the white pulp with very few c-Kit+ cells detected overall (Figure 2a). This result was not expected since adult murine spleen is known to harbour very low levels of Sca-1+c-Kit+ HSC under steady-state or normal physiological conditions [18, 19]. In contrast, antibody to c-Kit identified extensive distribution of c-Kit+ cells in the red pulp of neonatal spleen. Cells which showed co-staining with Sca-1 were potentially HSC (Figure 2b) while cells staining only with c-Kit could reflect haematopoietic progenitors/precursors or even dendritic cells. Small numbers of c-Kit+ cells were also found in the white pulp, and these could reflect dendritic cells (Figure 2b). Co-staining of Sca-1+c-Kit+ cells reflecting HSC in red pulp, along with a distinct population of c-Kit+ cells, was confirmed by high power microscopy (Figure2b’).
In order to increase the possibility of detecting HSC in spleen, mice were treated with Flt3L, a cytokine which has been shown to mobilise bone marrow cells into blood and spleen when administered to mice [23, 25]. Mice were given an inoculum of B16-Flt3L melanoma cells which secrete Flt3L [37] and after 7 days were sacrificed for quantitation of HSC present in spleen. Spleens were also stained using two staining protocols to detect HSC. The detection of Lin-Sca-1+c-Kit+ cells is known to detect a heterogeneous population of HSC [34, 39]. Staining for Lin-CD150+CD41-CD48- cells is an alternative, more specific stain for long-term (LT)-HSC, but this is also known to be a heterogeneous population [8]. Section staining of spleens from Flt3L-mobilised mice using antibodies to Sca-1 and c-Kit confirmed co-staining of these two markers on some spleen cells in the red pulp region consistent with the presence of HSC/HSPC (Figure 2c). Some cells in white pulp also stained only for cKit and these were possibly dendritic cells or other myeloid precursors (Figure 2c). Lineage staining of the same sections identified hematopoietic cells including lymphoid and myeloid cells in spleen (Figure 2d). However, these c-Kit expressing cells did not express lineage markers, a result also consistent with the hypothesis that HSC/HSPC have localised into spleen on mobilisation.
When the more definitive CD150 and CD41/CD48 staining was applied, spleens from Flt3L-mobilised mice revealed no clear population of CD150+CD41/48- cells identifiable as LT-HSC (Figure 2f). These were also not identifiable in normal adult mice (Figure 2e). Several large CD150+CD41/48+ cells were identified however and were also in much higher number in untreated adult spleen (Figure 2e). Based on their large size and phenotype these cells were classified as megakaryocytes which have previously been reported in adult spleen [40, 41].
Quantitation of HSC in spleen
Flow cytometry was used to quantitate HSC and HSPC subsets present in spleens of adult, neonatal, pregnant and Flt3L-mobilised adult mice. These studies confirmed the presence of rare primitive LT-HSC in spleens of pregnant mice in higher numbers than were detected in either neonatal mice or normal adult control mice. This study involved multicolour staining for initial detection of the rare Lin-Sca-1+c-Kit+ (LSK) subset in spleen, followed by gating to identify CD150+Flt3- cells which were then tested for expression of CD41/CD48 (Figure 3a). The CD150+Flt3-CD41-CD48- LT-HSC subset was quantified in spleens, along with other HSPC subsets delineated on the basis of differences in expression of these markers including short-term (ST)-HSC (Lin-Sca-1+c-Kit+Flt3-CD150-CD48-), multipotential progenitor subset 2 (MPP2) (Lin-Sca-1+c-Kit+Flt3-CD150+CD48+) and MPP3 (Lin-Sca-1+c-Kit+Flt3-CD150-CD48+) subsets [42, 43].
These studies indicated a significant ~15-fold increase in the size of the LT-HSC population in pregnant mice over normal adult spleen (p=0.018) and ~10-fold increase over neonatal spleen (p=0.0623; not significant) (Figure 3c). While apparently higher numbers of ST-HSC, MPP2 and MPP3 were detected in pregnant spleens, the numbers of these cell types were not significantly different from either adult spleen or neonatal spleen, except for MPP3 which were in significantly higher number in pregnant over adult mice (p=0.0304). Quite large variation was detected in the numbers detected in pregnant mice and this would appear to be a biological feature related to pregnancy in mice and its effect on spleen hematopoiesis.
In order to determine the specific subsets of HSPC which were increased in frequency in spleens of Flt3L-treated mice, a comparison was made of the relative numbers of LT-HSC, ST-HSC, MPP2 and MPP3. Figure 3b demonstrates the gating analysis used to identify these subsets in Flt3L-mobilised and control adult mice. These studies confirmed that the specific subset of MPP3 was present in significantly higher numbers in pregnant mice than LT-HSC, ST-HSC or MPP2 (Figure 3d). The numbers of LT-HSC, ST-HSC and MPP2 was similar, although the number of MPP3 was ~30-fold higher and significantly different than than the other subsets in pregnant mouse spleen (Figures 3c, d). MPP2 reflect megakaryocyte-biased progenitors while MPP3 are myeloid-biased progenitors [44]. Flt3L-mobilisation is therefore more selective for the MPP3 subset of myeloid progenitors than for HSC, in particular.
Localisation of HSC amongst spleen stromal environments
The different non-hematopoietic stromal cell types were then localised in spleen so that the stromal environment of HSC could be identified. Antibodies specific for cell surface markers were used to stain frozen sections including CD29, a marker of mesenchymal cells and some hematopoietic cells in spleen [26, 27], CD105 which marks angiogenic vasculature, marginal reticular cells and red pulp fibroblasts which appear in neonates ahead of full hematopoiesis [44], CD31 which identifies mature endothelial cells in red and white pulp of spleen [26, 29], gp38 which has been used to identify fibroblastic reticular cells and follicular dendritic cells in spleen [26, 45] [26, 30], CD140a/b which identifies perivascular reticular cells and mesenchymal stem cells [10, 26, 31], CD51 which identifies perivascular reticular cells [31, 46], and Thy1/CD90 which identifies T cells and mesenchymal stem/progenitor cells [31-34]. By using a combination of markers for stromal and hematopoietic cell types, we have been able to investigate the localisation of different stromal subtypes in relation to HSC in neonatal and pregnant mouse spleen.
Initial studies involved neonatal spleens which have a more disorganised stromal cell distribution than adult spleen (Figure 1 & Supplementary Figure S1). Sections were stained with CD150 and co-stained with CD41/CD48/Lin to identify CD150+CD41/48/Lin- LT-HSC. F4/80 was used as a co-stain to demarcate the red pulp region filled with myeloid cells, and then CD31, gp38 or CD105 were used as a co-stain to delineate different stromal cell types. Low power microscopy of sections stained with CD31and F4/80 were used to identify white pulp with the central CD31staining central arteriole and red pulp filled with F4/80 staining myeloid cells (Figure 4a). The high-power view identified CD150+CD41/48/Lin‑ LT-HSC in red pulp which are not specifically located in the vicinity of either F4/80+ myeloid cells or CD31+ vascular cells (Figure 4a’). Similar staining involving gp38 instead of CD31 identified a dense network of fibroblastic reticular cells in the red pulp, along with many Lin+ cells which demarcated the white pulp, and large, bright CD150+CD41/CD48+ megakaryocytes in the red pulp region (Figure 4b). High power microscopy identified CD150+CD41/48/Lin- LT-HSC in red pulp which appeared clustered amongst Lin+ cells. Some LT-HSC were located alongside gp38 stromal cells (Figure 4b’). A final staining involved addition of CD105 which stains predominantly sinusoids in red pulp and also demarcates central arterioles and marginal zones (Figure 4c). Localisation of CD150+CD41/48/Lin- cells under high power revealed again that LT-HSC were present in the red pulp region and not associated closely with CD105 staining sinusoids (Figure 4c’).
Further studies sought to localise LT-HSC in pregnant spleens. Staining involved detection of CD150+CD41/48/Lin- cells in relation to F4/80+ myeloid cells demarcating the red pulp and CD3 demarcating the white pulp (Figure 5a). Pregnant adult spleen has much more pronounced regions of red pulp, and white pulp comprising T and B cell areas. A high-power view clearly localised LT-HSC in the red pulp, with cells clearly scattered in proximity with F4/80 staining myeloid cells (Figure 5a’). Co-staining for CD31 clearly identified white pulp regions with a central arteriole, and other vasculature scattered across the red pulp (Figure 5c). A high-power view clearly distinguished a small number of LT-HSC, but these were not associated with CD31 staining endothelial cells (Figure 5c’). Co-staining for Thy1.2 /CD90 clearly stained T cells in the white pulp which was devoid of LT-HSC, and also showed scattered mesenchymal stem/progenitor cell staining in the red pulp (Figure 5d). A high-power view identified LT-HSC scattered amongst the myeloid cells in red pulp, with a small number in association with Thy1.2/CD90+ mesenchymal stem/progenitor cells (Figure 5d’). Further staining involved costaining for gp38 which clearly detects fibroblastic reticular cells in the white pulp (Figure 5e) in association with mature Lin+ cells (Figure 5e’’). Antibody to gp38 gave no staining of cells in the red pulp (Figure 5e’), although LT-HSC were detectable in clusters amongst the myeloid cells in red pulp (Figure 5e). A final staining involved analysis of pregnant mouse spleen sections for CD140b, a marker which identifies mesenchymal perivascular reticular cells in red pulp, and in white pulp, a central clump of CD140b stained cells which represent follicular dendritic cells [47]. Co-staining with c-Kit was used to identify HSPC in spleen and to determine any association with CD140b-staining cells. Figures 5 (f, g, h) identify c-Kit staining cells in red pulp, some of which co-stain with CD140b, suggesting close proximity between HSPC and perivascular reticular cells. Scattered c-Kit staining cells in the white pulp could reflect dendritic cells which do not co-stain with CD140b (Figure 5g). Also, some cells in the red pulp do not co-stain with c-Kit suggesting that only a subset of CD140b staining cells in the red pulp are localised in close proximity with HSPC, perhaps reflective of the stem cell niche.
In summary, some although not all HSC in neonatal mice appeared to associate with gp38+ (% association: 32.50 ± 22.98) or with Lin+ cells (Figure 4 b, b’). In contrast, in pregnant spleens, HSC appeared to associate with F4/80+ myeloid cells or Lin+ cells in the red pulp of spleen, in the presence of far fewer gp38+ cells (Figure 5 e, e’). A final study involving Thy1.2 staining and localisation of cells in red pulp of pregnant mouse spleen, revealed that some HSC in these mice were in close alignment with Thy1.2 staining mesenchymal cells (% association: 26.17 ± 15.11), as well as F4/80 and Lin staining cells (Figure 5 d, d’). Thy1.2 staining would be consistent with the presence of mesenchymal perisinusoidal cells providing a niche in spleen supportive of HSC as reported by others [10].
Identification of cells which form hematopoietic niches in adult spleen
Very little is known about the subsets of CD140-expressing mesenchymal cell subsets which populate spleen, although their importance as niche elements for HSC has been recently described [10, 15, 48, 49]. Since pregnant spleens contain quantifiable numbers of LT-HSC (Figure 3c), it was reasoned that adult spleens contain niches for HSC, and that adult spleen would be a more informative model to study stromal niches than neonates. The stromal fraction of adult mouse spleen was therefore isolated and sorted to isolate any CD140a/b expressing cells. Single cell sorting was used to isolate cells for preparation of RNA, and subsequent deposition into a Fluidigm chip. The presence of live cells was verified microscopically, and cDNA prepared for each cell in the wells of the chip. RNA library preparation and sequencing led to identification of gene expression for each cell.
Initial analysis involved RNA sequencing of 36 isolated cells, but initial screening of gene expression identified 12 cells as non-viable with high expression of mitochondrial genes. Data analysis proceeded by clustering on the basis of gene expression in these 24 cells. A first approach to subset identification involved selection of genes for clustering which had the highest variance across cells. Genes were only included in the analysis if they had signal value >100 in every cell. Initial analysis involved clustering of 8000 most variable genes, and this number was reduced until clear clustering was observed. Clustering of the 80 most variable genes was most informative, since it revealed clear separation into three clusters of cells (Figure 1c). A second approach involved selecting the highest expressed genes for clustering and involved setting different cut-offs for gene expression. The criterion for gene selection was that at least one cell must express the gene > cut-off signal value. For example, cut-off 100 gave 7515 genes for clustering, cut-off 1000 gave 6149 genes, cut-off 10,000 gave 2218 genes, and cut-off 100,000 gave 27 genes. Cut-off 1000 was selected since it gave 3 clear clusters which mirrored the same 3 clusters of cells identified on the basis of clustering the 80 most variable genes (Figure 6b). Only one discrepancy was evident whereby C82 was in cluster 3 in the first analysis involving the 80 most variable genes, but in cluster 3 in the second analysis involving signal value cut-off of 1000. The outcome of finding three clear clusters suggests 3 possible cell subsets are present amongst the CD140a/b expressing cells.
Data mining was then used to investigate the expression of genes reflecting distinct cell lineages based on knowledge of CD140a/b expression on cells. Initial studies showed an absence of genes reflective of common stromal cell types including endothelial cells, and smooth muscle cells or pericytes. Since CD140a and CD140b are known markers of several stromal cell subsets including mesenchymal stem cells (CD140a/b+), perivascular reticular cells (CD140b+) and follicular dendritic cells (CD140a+), sets of genes reflective of these three main lineages were compiled through literature searching. Gene expression values were ranked for each of the three cell clusters identified previously. These data showed clear delineation of the clusters into different cell type. Cluster 1 cells expressed high levels of genes most reflective of follicular dendritic cells including Cxcl13, Mfge8, Acta2 and Ccl19 which was not expressed by cells in clusters 2 and 3 (Figure 6d). Cluster 1 cells also expressed high levels of many genes expressed by mature mesenchymal cells, also consistent with the classification of follicular dendritic cells. These genes included Col1a1, Igfbp3, Itgb1, Vcam1, Sept4, Lepr, Cd44, Pdgfrb, Bmpr2, Itga1, Itgav, Cd164 and Pdgfra (Figure 6d). Clusters 2 and 3 reflect more closely aligned cells, although Cluster 2 cells are distinguishable by lower expression levels of genes expressed by both mesenchymal and perivascular cells. The consideration is that Cluster 2 cells could reflect more immature mesenchymal or perivascular reticular cells than those in Cluster 3. Cluster 3 cells are characterised by expression of genes known to be highly expressed in perivascular reticular cells notably Cxcl12, KitL (SCF), Hmcn1, Tcf21, Ctnnb1, Alcam and Tlx1 (Figure 6d). These cells also expressed high levels of genes expressed by mature mesenchymal cells as identified for Cluster 1 cells, reflective of cells of the mesenchymal lineage but of a different functional type.
Single cell studies using the Fluidigm technology represent a powerful tool to identify cell subsets and their gene profile. These data confirm the presence of CD140a/b follicular dendritic cells in spleen which were also detectable as a central clump in the B cell follicles, through immunocytochemical staining of spleen white pulp (Figure 5g & h). Gene expression data also confirms the presence of 2 clusters of CD140b-expressing cells subsets corresponding to perivascular reticular cells, which reflect those located in the red pulp of spleen by immunocytochemical staining (Figure 5g & h), only some of which colocalise with c-Kit staining hematopoietic progenitor cells.
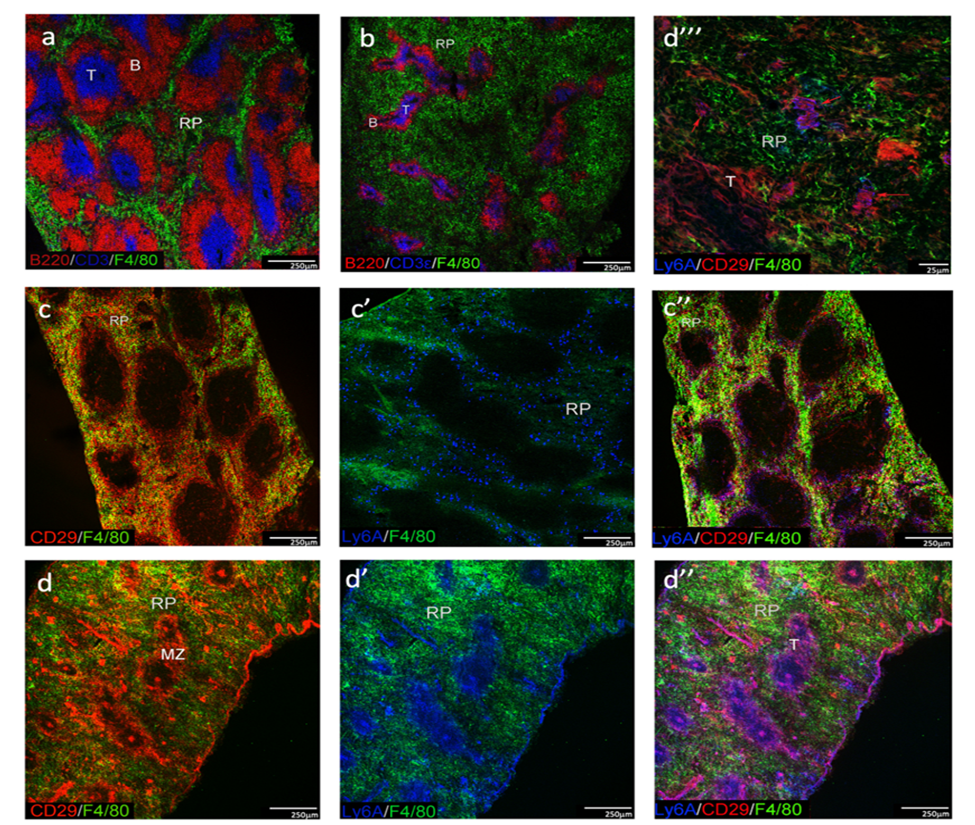
Fig 1: Comparison of spleen morphology between adult and neonates
Spleens were collected from D6 neonatal and adult W6 mice. Acetone-fixed frozen sections were stained with specific fluorochrome-conjugated antibodies and representative images are shown for CD3, B220 and F4/80 staining of T cell zones (T), B cell follicles (B) and red pulp (RP) in both adults (a) and neonates (b). Scale bar: 250mm. Spleen stroma was investigated through staining of CD29 and Sca-1, using F4/80 to delineate myeloid cells in RP of adults (c, c’, c’’) and neonates (d, d’ d’’). Scale bar: 250mm (c, d). High power view of neonatal spleen (d’’’) indicates CD29+Sca-1+/- stromal cells in RP (red arrows). Scale bar: 25mm. Images representative of 3 mice are shown.
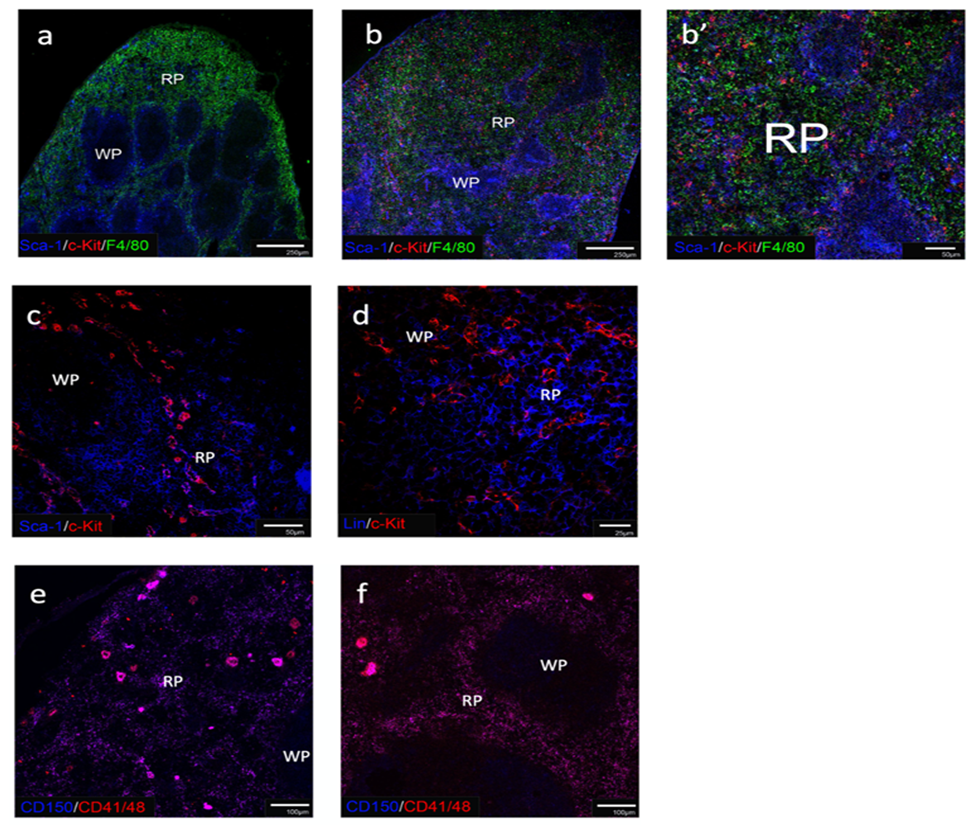
Fig 2: Detection of HSPC in spleens of neonates and adults
In order to detect HSPC is spleen, acetone-fixed frozen sections were stained with specific fluorochrome-conjugated antibodies. Adult (a) and neonatal (b, b’) spleen were investigated for Sca-1 and c-Kit staining to detect cKit+Sca-1+ HSPC. F4/80 staining was used to detect myeloid cells and so delineate red pulp (RP) from white pulp (WP). Scale bar: 250μm) (a, b) and 50mm (b’). Spleen of adult mouse treated with Flt3L was stained for Sca-1 and c-Kit (c) and Lin and c-Kit (d) to detect HSPC as cKit+Sca-1+ cells but not Lin+c-Kit+ cells. Scale bar: 50mm (c) and 25 mm (d). Spleens of adult normal mice (e) were compared with Flt3L treated mice (f) for CD150, CD41 and CD48 staining to identify any CD150+CD41/48- HSPC. Scale bar: 100mm. Images representative of 3 mice are shown.
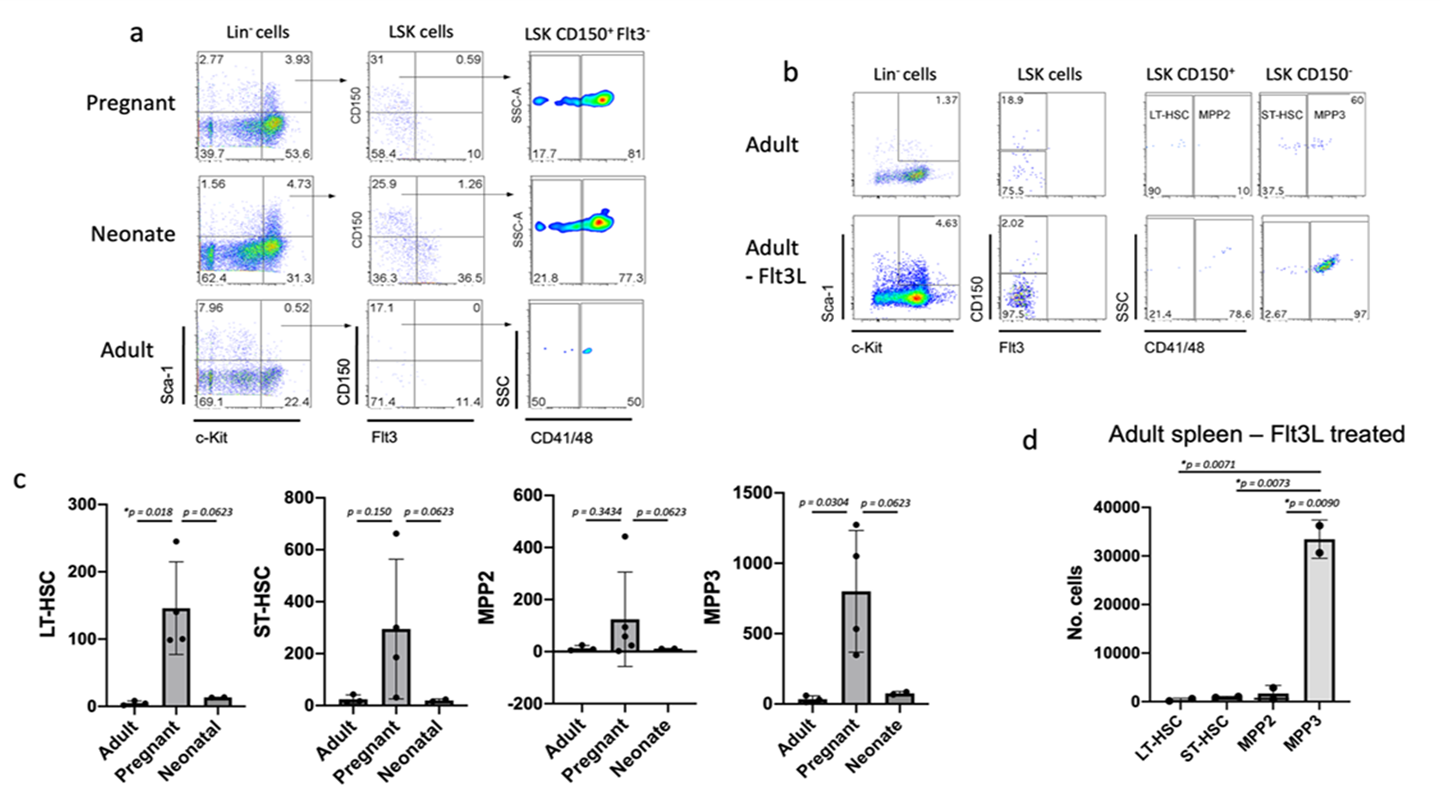
Fig 3: Flow cytometric detection of HSPC in spleen
(a) Spleen cells from pregnant (E18.5), neonatal (D6) and adult mice were subjected to magnetic bead cell separation to deplete Lin+ cells and then labelled with antibodies specific for Lin and the markers Sca-1, c-Kit, CD150, Flt3, CD41 and CD48 to gate LT-HSC as a Lin-Sca-1+c-Kit+CD150+Flt3-CD41-CD48- subset. (b) Spleen cells from Flt3L-mobilised and normal adult mice were stained as in (a) and gated to identify LT-HSC (Lin-Sca-1+c-Kit+Flt3-CD150+CD48-), ST-HSC (Lin-Sca-1+c-Kit+Flt3-CD150-CD48-), MPP2 (Lin-Sca-1+c-Kit+Flt3-CD150+CD48+) and MPP3 (Lin-Sca-1+c-Kit+Flt3-CD150-CD48+) subsets. (c) Absolute number of each HSPC subset identified in spleens of adult (W6), pregnant and neonatal (D6) mice (E18). (d) Absolute number of HSPC subsets present in spleen of Flt3L-treated adult mice. Data are shown as mean + SE (n=3). * significantly different (p less than0.05)..
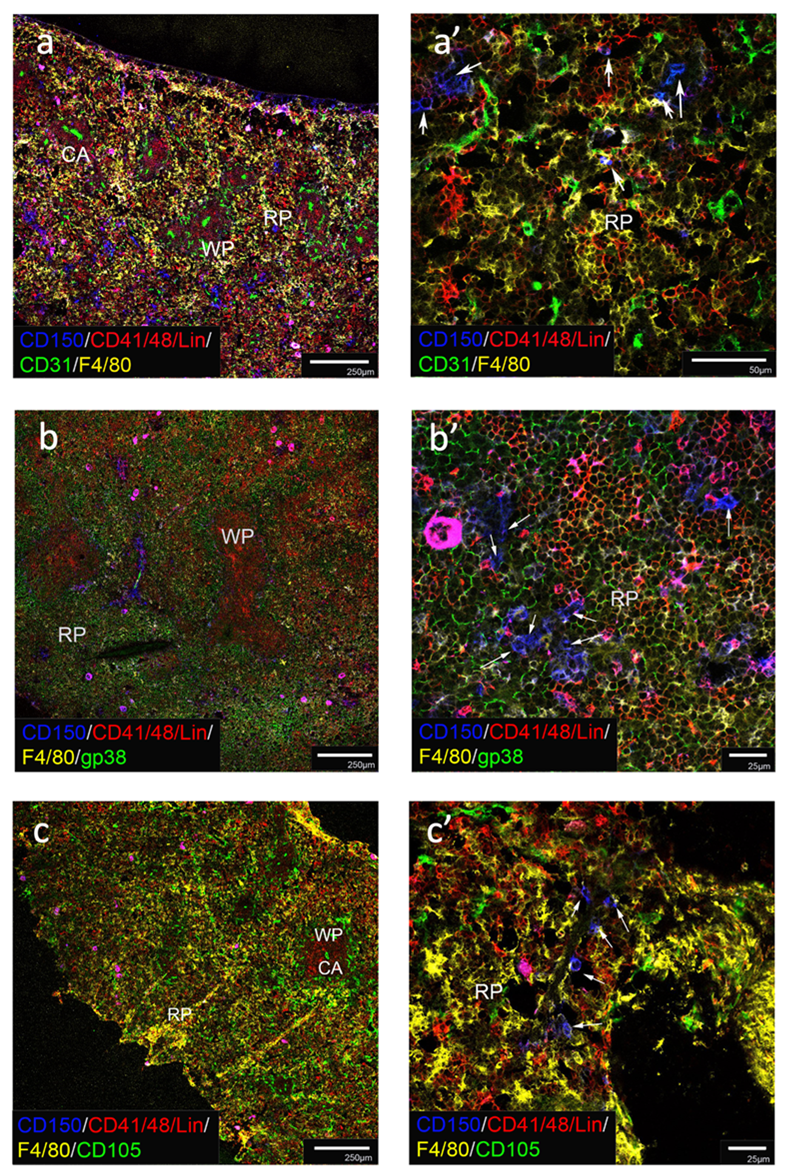
Fig 4: Localisation of HSC in neonatal spleen
Acetone-fixed frozen neonatal spleen sections were stained with fluorochrome-conjugated antibodies specific to CD150, CD41, CD48 and Lin to identify HSC as CD150+CD41/48-Lin- cells along with antibodies to F4/80 (staining myeloid cells in red pulp) and either CD31 (a, a’), gp38 (b, b’) or CD105 (c, c’) to assess HSC association with different stromal cell types. Red pulp (RP), white pulp (WP) and central arteriole (CA) regions are identified. White arrows identify HSC (a’, b’, c’). Scale bar: 250μm (a, b, c) 50μm (a’, b’, c’). Images representative of 3 mice are shown.
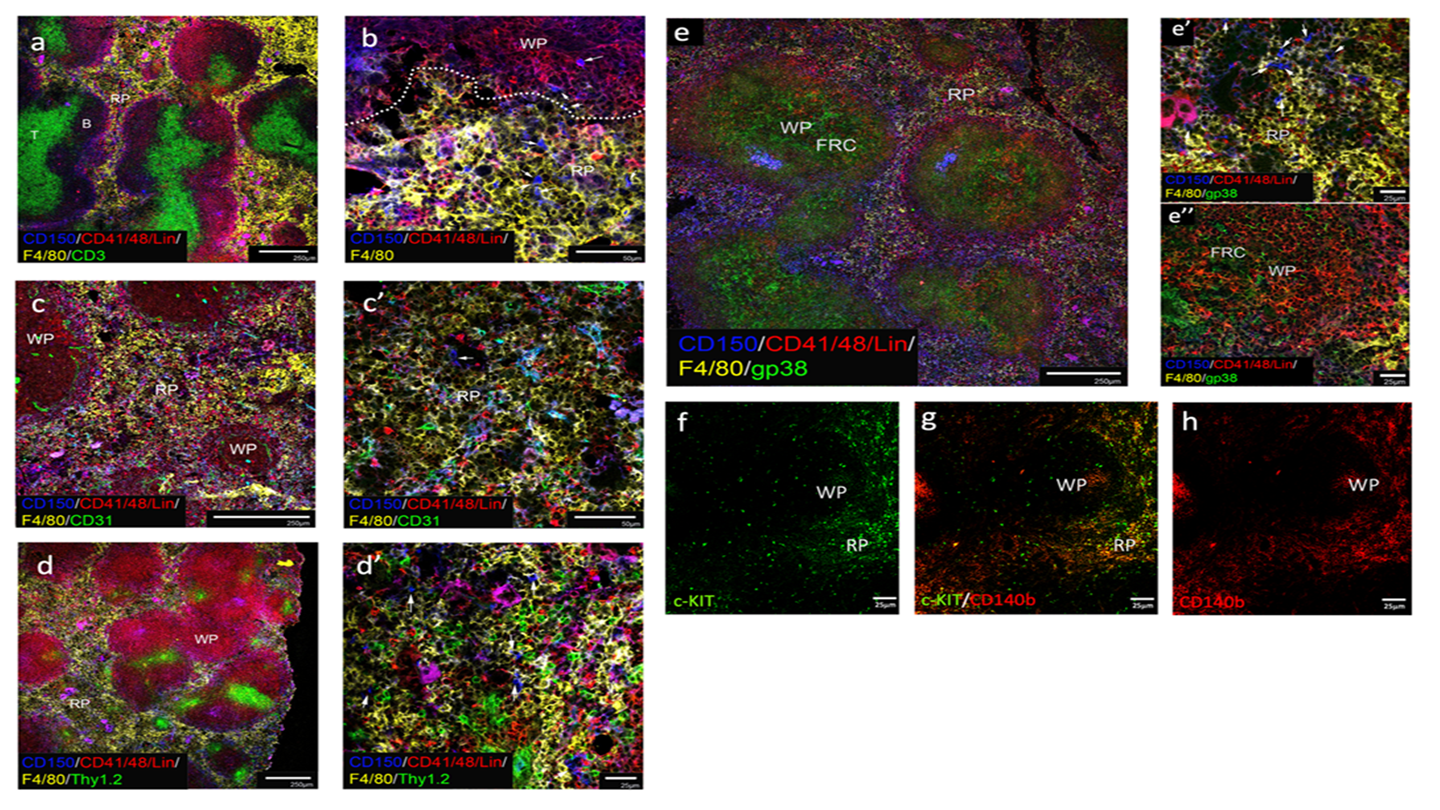
Fig 5: Localisation of HSC in pregnant spleen
Acetone-fixed frozen spleen sections from adult pregnant (E18.5) mice were stained with fluorochrome-conjugated antibodies specific to CD150, CD41, CD48 and Lin to identify HSC as CD150+CD41/48-Lin- cells, along with antibodies to F4/80 to delineate myeloid cells in red pulp. Other antibodies used were specific for CD3 (a), CD31 (c, c’), Thy1.2 (d,d’) and gp38 (e, e’, e’’) to assess HSC association with different cell types. Red pulp (RP), white pulp (WP), T cell areas (T), B cell areas (B) and fibroblastic reticular cells (FRC) are identified. White arrows identify HSC (b, c’, d’, e’). c-Kit+ cells localised in RP were localised in association with CD140b+ stromal cells (f, g, h). Scale bar: 250μm (a, c, d, e), 50μm (b, c’,), 25μm (d’, e’, e’’, f, g, h). Images representative of 3 mice are shown.
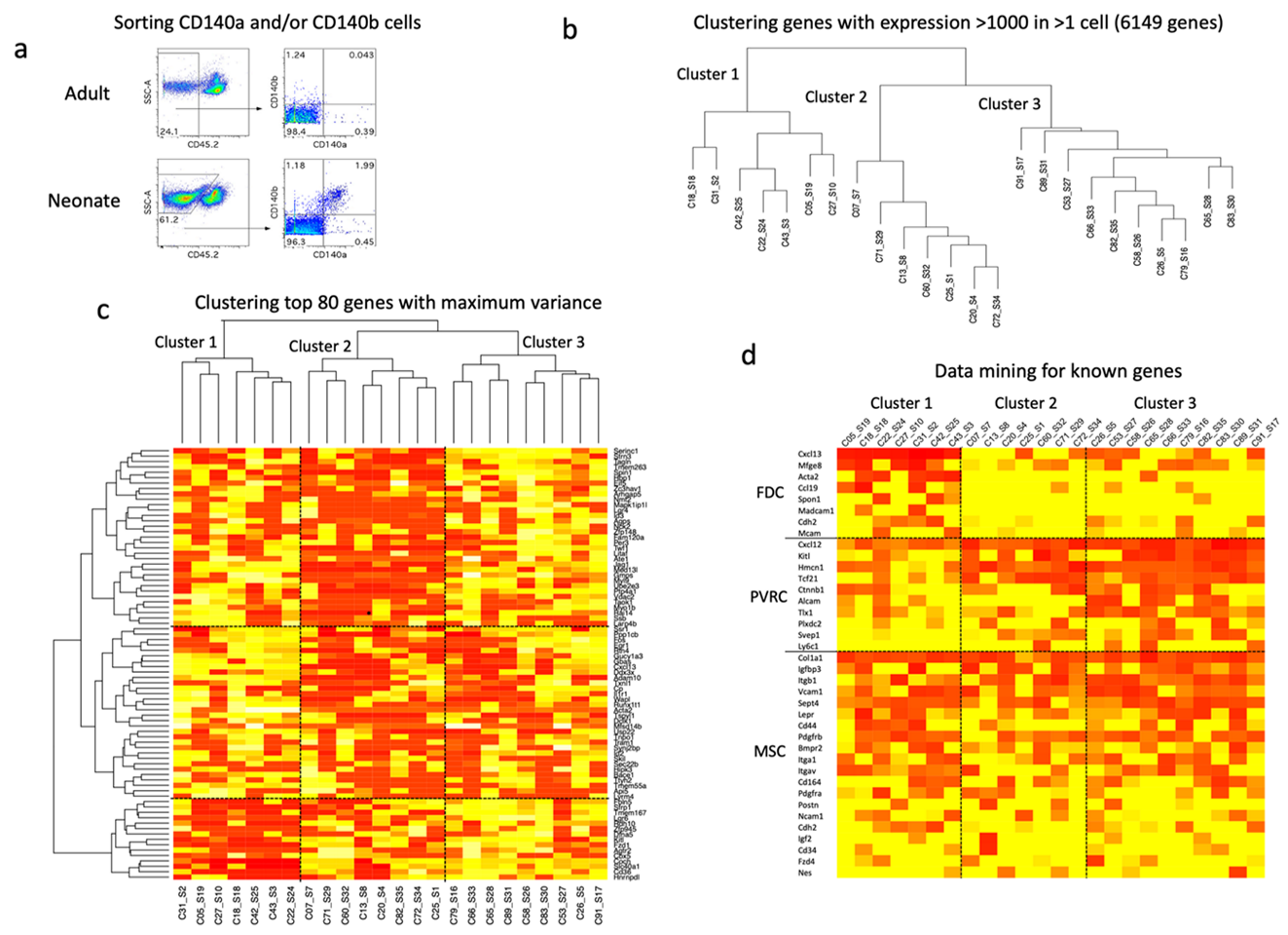
Fig 6: Characterisation of HSC niches in neonatal spleen
Gene expression analysis through single cell sequencing employed the C1 Fluidigm system to investigate the CD140a/b+ subset of splenic stroma. (a) Initial analysis compared the number of stromal cells which could be isolated from adult W6 and neonatal mice D6 through cell sorting to gate out CD45.2+ lymphoid cells for the identification of CD140a/b+ cells for sorting. Single cell deposition, library production and sequencing were performed on CD140a/b+ cells sorted from neonatal spleen. Twenty-four cells were selected for analysis. (b) Hierarchical clustering of highly expressed genes is shown for 6149 genes expressed at a signal value >1000 in at least one cell, giving three main clusters. (c) Hierarchical clustering of the 80 most variable genes also revealed the same three main clusters. (d) Data mining was used to detect expression of known genes by these clusters. Genes were selected known to be expressed in perivascular reticular cells (PVRC), mesenchymal stem cells (MSC) and follicular dendritic cells (FDC). Genes expressed at signal levels of >1000 in more than one cell are shown in the heat map. Heat maps show ln expression for each cell across the signal level of 0 (red) to maximum signal (yellow).
Discussion
While the concept of an HSC niche was developed a few decades ago, it has only been recent technological advances which have permitted experimental investigation at the level of single cell identification and interaction. Indeed, those studies have uncovered multiple niche elements in bone marrow and described the possible interplay between distinct cell types. Unfortunately, investigation into HSC niches in spleen is only in its infancy and made more difficult because of the rarity of HSC in that organ. Information on the cell surface markers of splenic stroma has been used here to investigate niches for HSC in spleen.
Previous studies have shown that the adult spleen contains a very small population of HSC capable of long-term hematopoietic reconstitution [18, 50]. HSC in spleen were also found to be phenotypically and functionally similar to HSC in the bone marrow [38]. However, since HSC exist in far lower number in the adult spleen than that in the bone marrow [38], it has been extremely challenging to detect HSC in steady-state adult spleen using immunofluorescence imaging. We have therefore tested animal models where the numbers of HSC might be expected to increase above normal adult levels. These included neonatal mice (D6), pregnant (E18.5) mice and Flt3L treated mobilised mice. There are two main reasons for the choice of the neonatal spleen model. Firstly, it is known that the spleen harbours high number of HSC until 2 weeks of age [18, 51]. Secondly, splenic long-term cultures were most readily established from neonatal D6 spleen, producing myeloid dendritic-like cells which develop directly from LT-HSC [52]. The choice of the pregnant model is largely based on a recent report which showed that pregnant adult spleen contains more HSC than non-pregnant adult spleen, along with increased erythropoiesis and myelopoiesis [10]. In addition, the documented effect of Flt3L on myeloablation and HSC mobilisation from bone marrow to peripheral blood and spleen [23-25], was the main reason for the choice of Flt3L mobilisation model.
In this study, we did localise Lin-CD150+CD41-CD48- LT-HSC in the red pulp of neonatal spleen by section staining, although they were present in low numbers. Our data therefore parallel a recent report showing that HSC in spleen are localised in the red pulp following induction of extramedullary hematopoiesis [10]. In that report, extramedullary hematopoiesis was induced in spleen using combined administration of cyclophosphamide and G-CSF which results in increased numbers of HSPC migrating into spleen and allowed detection of HSC in spleen by immunofluorescence imaging [8, 10, 53, 54]. The same group also showed that extramedullary hematopoiesis was prominent in spleens of pregnant mice [22]. Consistent with their findings, we found that the number of Lin-CD150+CD41-CD48- LT-HSC in pregnant adult mice was significantly greater by ~15-fold than in normal adult spleen (Figure 3c). In addition, Lin-CD150+CD41-CD48- LT-HSC were located in the red pulp of pregnant spleens (Figure 5). These results are consistent with increased erythropoiesis and myelopoiesis in spleen during pregnancy dependent on an increased supply of HSPC to that organ [10, 22]. In contrast, extramedullary hematopoiesis in neonatal spleen progressively decreases to a steady-state level during development to adulthood as the bone marrow takes over as the main hematopoietic organ. The same population of LT-HSC is in far lower in number in Flt3L mobilised spleens compared with control adult spleens. It has been shown in our study that Flt3L treatment resulted in high numbers of MPP3, a myeloid biased MPP in the spleen (Figure 3d). However, it is unclear if the differentiation of HSC to MPP occurred prior to mobilisation, or within the splenic microenvironment.
This study also looked at the stromal cells which lay in association with HSC as a first attempt to describe stromal niches in spleen. Multiple cell surface markers including CD31, gp38, CD105 and Thy1.2 were used in this study. Amongst those tested, no association was observed with CD31 and CD105 cells in either neonatal or pregnant spleens (Figures 4 & 5). Staining for CD105, a marker of angiogenic vasculature, marginal reticular cells and red pulp fibroblasts, gave a similar staining pattern to CD31 in both pregnant adult and neonatal spleens (Figure 3B). However, CD105 staining in red pulp (F4/80+) of neonatal spleen was stronger than in pregnant adult spleens, consistent with a predominance of CD105+ stromal cells during neonatal development [55].
In neonatal spleens which have a predominance of gp38+ stromal cells in red pulp, some LT-HSC appeared to associate with gp38+ stromal cells (Figure 4b, b’). This however was not the case in adult pregnant spleen where fewer gp38+ cells can be found and very few LT-HSC appeared to colocalise with these cells (Figure 5e, e’ e’’). Thy1.2 staining stromal cells did however appear to lie in close proximity with some LT-HSC in pregnant spleens (Figure 5 d, d’), consistent with a niche involving mesenchymal, perhaps perisinusoidal reticular cells as described previously by others [10].
It was also noted that some LT-HSC were associated with Lin+ hematopoietic cells or myeloid cells. It is possible that these cells act to maintain LT-HSC within the splenic red pulp micro-environment which is otherwise inhospitable, owing to the highly immunoreactive nature of the organ. Consistent with this finding is the reported role of bone marrow CD169+ macrophages in HSC maintenance [56], so that splenic macrophages may also promote HSC maintenance. Indeed, Dutta and co-workers recently reported that macrophages retain HSC in spleen using VCAM1, and it was found that reduction of VCAM1 expression leads to egression of HSC into the circulation [57]. Tang and co-workers also demonstrated that splenic stroma promotes differentiation of bone marrow HSC into regulatory dendritic cells [58]. It is interesting to speculate that some of these Lin+ cells are regulatory dendritic cells, which themselves promote the generation of regulatory T cells. Recently, FoxP3 regulatory T cells were reported to provide an immune-privileged site for the HSC niche in bone marrow [59]. It is possible that these regulatory T cells reside in close proximity to HSC to protect them from the immune environment in spleen. Indeed, several immune cell types might be involved in the support and regulation of HSC in spleen.
Although direct contact of HSC with stromal cells is in keeping with a niche model for hematopoiesis, it is still possible that stromal cells could act in a paracrine fashion. We are therefore unable to conclusively interpret the hematopoietic support role of stromal cells which were not in direct contact with HSC without using a conditional knock out mouse model to undertake a study. A splenic niche involving Tcf21+ stromal cells has been recently reported [10]. Tcf21+ cells were shown to be located around sinusoids in red pulp of spleen and most HSC were found to be associated with those cells [10]. The production of SCF and CXCL12 by Tcf+ stromal cells, and their important role in extramedullary hematopoiesis, was demonstrated in knock-out mouse models [10]. That study identified Tcf21-expressingstroma as CD140b+ perivascular cells in spleen red pulp as we have shown here. Our study also shows high levels of expression of Scf and Cxcl12 by cells expressing Tcf21 (Figure 6d). They were unable to show that some CD140b+ cells like the follicular dendritic cells identified here do not express Tcf21 and are unlikely to be HSC niches.These experiments are an insight into splenic niches, but not a full characterisation of stromal subsets and their capacity to support hematopoiesis.
Distinct architectural differences were also noted between adult and neonatal spleens in this study. The more developed white pulp region of the adult spleen as compared to the neonatal spleen is consistent with earlier findings that the white pulp of murine spleen is poorly organised at birth and that extensive cellular organisation only occurs after birth [60, 61]. This greatly contrasts with the human spleen which is apparently fully developed at birth [62]. To this end, we have shown that HSC localise in the red pulp of neonatal and pregnant spleens. While only gp38+ stromal cells were found to associate with HSC in neonatal spleens, there appear to be mesenchymal stromal cell types which form a niche in adult spleen, with the possibility that endothelial and vascular cells may contribute in a paracrine fashion to aid in the regulation of splenic extramedullary hematopoiesis.
Declarations
Acknowledgements
Statistical analysis and clustering performed by Professor Terence O’Neill (Bond Business School, Gold Coast, QLD, Australia) is recognised with gratitude.
Data Availability Statement
All original data is included in this publication. The datasets used and/or analysed supporting the conclusions of the current study are available from the corresponding author on reasonable request: honeill@bond.edu.au.
Ethics Statement
The animal study was conducted according to protocols approved by the Animal Experimentation Ethics Committee at the Australian National University (ANU: Canberra, Australia) (protocol number: #A2013/11).
Author Contributions
HKL: Investigation, Methodology, Data curation, Data analysis, Writing original draft, Writing – review and editing. HCO: Data analysis, Supervision, Writing – review and editing.
Funding Information
This project was supported by project grants to HO from the Australian Research Council (#DP13010307) and the National Health and Medical Research Council of Australia (#585443). HL was supported by an Australian National University Postgraduate Scholarship.
Conflict of Interest Statement
The authors declare that they have no competing interests.
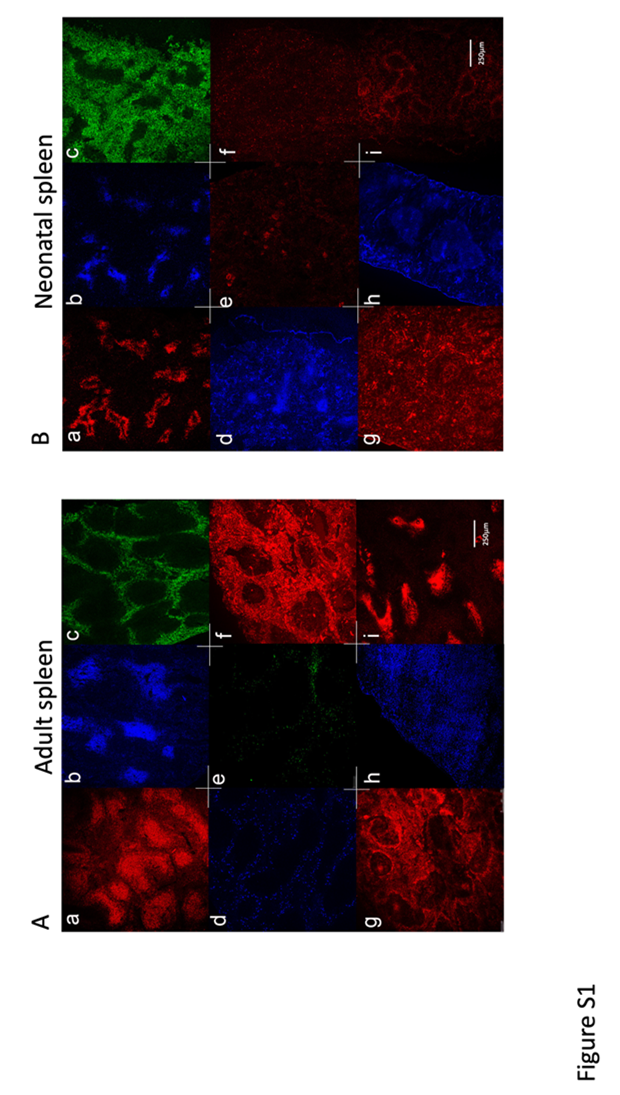
Fig: S1
Distinct stromal cell distribution in neonatal compared with adult spleen
Initially, spleen cells expressing various markers of hematopoietic and stromal cells were localised in single colour studies using specific antibodies (Figure S1). Adult 6-week and neonatal D6 spleens were compared for staining of markers reflecting T cells (CD3e), B cells (B220) and myeloid cells (F4/80). These stains revealed the distinctive T cell zones and adjacent B cell follicles which make up the white pulp regions. F4/80 staining of myeloid cells demarcated the red pulp region of spleen. Neonatal D6 spleens had notably larger red pulp regions and smaller white pulp regions for T and B cells (Figure S1B a, b, c), while these areas were larger and more demarcated in adult spleen (Figure S1A a, b, c). The more organised architecture of the splenic stroma in adult compared with neonatal spleen is identified through staining with CD29 and CD54 (Figure S1A d, g; Figure S1B g). Staining adult spleen for CD45.2 revealed high numbers of lymphoid cells in the follicles as well as myeloid and lymphoid cells migrating through the red pulp (Figure S1A h). Staining for c-Kit, a receptor for stem cell factor (SCF), was used to detect HSPC as well as mesenchymal progenitors and stromal cells. While c-Kit staining was restricted to the red pulp region in adult spleen consistent with staining of HSPC and red pulp stroma (Figure S1A e), it was more evenly spread over all regions in neonatal spleen consistent with the presence of HSPC, as well as developing mesenchymal cells across all regions (Figure S1B f). Staining for CD150 in neonatal spleen was used to confirm HSC staining, and cells were detected predominantly in the red pulp region (Figure S1B e). CD150 staining revealed no cells in adult spleen (see Figure 3). Spleen sections were also stained for CD29, a β-1 integrin, expressed by red pulp fibroblasts and fibroblast reticular cells in the T cell zones, and also in the region of the central arteriole (Figure S1A f). Neonatal spleen staining for CD29 was distinctly different to adult spleen, with scattered clusters of CD29+ cells in the red pulp region, high numbers of CD29+ cells in the marginal zone and the T cell areas, and in periarteriolar regions (Figure S1B g). Antibody to Sca-1/Ly6A, a marker expressed by activated lymphocytes, HSPC and mesenchymal stem cells, clearly stained adult spleen cells on the edge of the follicles, or in the red pulp region of adult spleen (Figure S1A d). Conversely, Sca-1 staining cells in neonatal spleen were localised in the white pulp, with scattered clusters of Sca-1+ cells in the red-pulp (Figure S1B h). The general stromal markers Sca-1, CD54 and CD29 serve to demonstrate clear structural differences between neonatal and adult spleen. Adult spleen sections were densely stained by antibody specific for CD54 (ICAM1), with highest staining in the red-pulp, marginal zone, and region of central arteriole in the white pulp, consistent with evidence that CD54 is expressed by fibroblast reticular cells, follicular dendritic cells, marginal zone reticular cells and red pulp fibroblasts (Figure S1A g). Staining for CD140a (PDGFα) in neonatal spleen was found predominantly in the marginal zone on marginal zone reticular cells, with evidence for staining of fibroblast reticular cells in T cell zones as well as red pulp fibroblasts (Figure S1B i). Staining for gp38 (podoplanin) in adult spleens identified cells distributed in clusters, in the T cell zones reflecting FRC (Figure S1Ai).
References
- Boulais, P. E., & Frenette, P. S. (2015). Making sense of hematopoietic stem cell niches. Blood, The Journal of the American Society of Hematology, 125(17), 2621-2629.
- Ugarte, F., & Forsberg, E. C. (2013). Haematopoietic stem cell niches: new insights inspire new questions. The EMBO journal, 32(19), 2535-2547.
- Ding, L., Saunders, T. L., Enikolopov, G., & Morrison, S. J. (2012). Endothelial and perivascular cells maintain haematopoietic stem cells. Nature, 481(7382), 457-462.
- Méndez-Ferrer, S., Michurina, T. V., Ferraro, F., Mazloom, A. R., MacArthur, B. D., Lira, S. A., ... & Frenette, P. S. (2010). Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. nature, 466(7308), 829-834.
- Sugiyama, T., Kohara, H., Noda, M., & Nagasawa, T. (2006). Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity, 25(6), 977-988.
- Nilsson, S. K., Johnston, H. M., Whitty, G. A., Williams, B., Webb, R. J., Denhardt, D. T., ... & Haylock, D. N. (2005). Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood, 106(4), 1232-1239.
- Calvi, L. M., Adams, G. B., Weibrecht, K. W., Weber, J. M., Olson, D. P., Knight, M. C., ... & Scadden, D. T. (2003). Osteoblastic cells regulate the haematopoietic stem cell niche. Nature, 425(6960), 841-846.
- Kiel, M. J., Yilmaz, Ö. H., Iwashita, T., Yilmaz, O. H., Terhorst, C., & Morrison, S. J. (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. cell, 121(7), 1109-1121.
- Ding, L., & Morrison, S. J. (2013). Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature, 495(7440), 231-235.
- Inra, C. N., Zhou, B. O., Acar, M., Murphy, M. M., Richardson, J., Zhao, Z., & Morrison, S. J. (2015). A perisinusoidal niche for extramedullary haematopoiesis in the spleen. Nature, 527 (7579), 466-471.
- Crane, G. M., Jeffery, E., & Morrison, S. J. (2017). Adult haematopoietic stem cell niches. Nature Reviews Immunology, 17(9), 573-590.
- Periasamy, P., & O'Neill, H. C. (2013). Stroma-dependent development of two dendritic-like cell types with distinct antigen presenting capability. Experimental Hematology, 41(3), 281-292.
- Periasamy, P., Petvises, S., & O’Neill, H. C. (2013). Development of two distinct dendritic-like APCs in the context of splenic stroma. Frontiers in immunology, 4, 73.
- Periasamy, P., Tan, J. K., Griffiths, K. L., & O'Neill, H. C. (2009). Splenic stromal niches support hematopoiesis of dendritic-like cells from precursors in bone marrow and spleen. Experimental hematology, 37 (9), 1060-1071.
- O’Neill, H. C., Lim, H. K., Periasamy, P., Kumarappan, L., Tan, J. K., & O’Neill, T. J. (2019). Transplanted spleen stromal cells with osteogenic potential support ectopic myelopoiesis. PLoS One, 14(10), e0223416.
- Kim, C. H. (2010). Homeostatic and pathogenic extramedullary hematopoiesis. Journal of blood medicine, 13-19.
- Massberg, S., Schaerli, P., Knezevic-Maramica, I., Köllnberger, M., Tubo, N., Moseman, E. A., ... & von Andrian, U. H. (2007). Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell, 131(5), 994-1008.
- Wolber, F. M., Leonard, E., Michael, S., Orschell-Traycoff, C. M., Yoder, M. C., & Srour, E. F. (2002). Roles of spleen and liver in development of the murine hematopoietic system. Experimental hematology, 30 (9), 1010-1019.
- Dor, F. J., Ramirez, M. L., Parmar, K., Altman, E. L., Huang, C. A., Down, J. D., & Cooper, D. K. (2006). Primitive hematopoietic cell populations reside in the spleen: Studies in the pig, baboon, and human. Experimental hematology, 34(11), 1573-1582.
- Tan, J. K., & O’Neill, H. C. (2012). Myelopoiesis in spleen‐producing distinct dendritic‐like cells. Journal of Cellular and Molecular Medicine, 16(8), 1924-1933.
- Tan, J. K., Periasamy, P., & O'Neill, H. C. (2010). Delineation of precursors in murine spleen that develop in contact with splenic endothelium to give novel dendritic-like cells. Blood, The Journal of the American Society of Hematology, 115(18), 3678-3685.
- Nakada, D., Oguro, H., Levi, B. P., Ryan, N., Kitano, A., Saitoh, Y., ... & Morrison, S. J. (2014). Oestrogen increases haematopoietic stem-cell self-renewal in females and during pregnancy. Nature, 505 (7484), 555-558.
- Robinson, S., Mosley, R. L., Parajuli, P., Pisarev, V., Sublet, J., Ulrich, A., & Talmadge, J. (2000). Comparison of the hematopoietic activity of flt-3 ligand and granulocyte-macrophage colony-stimulating factor acting alone or in combination. Journal of Hematotherapy & Stem Cell Research, 9(5), 711-720.
- Neipp, M., Zorina, T., Domenick, M. A., Exner, B. G., & Ildstad, S. T. (1998). Effect of FLT3 ligand and granulocyte colony-stimulating factor on expansion and mobilization of facilitating cells and hematopoietic stem cells in mice: kinetics and repopulating potential. Blood, The Journal of the American Society of Hematology, 92(9), 3177-3188.
- Brasel, K., McKenna, H. J., Morrissey, P. J., Charrier, K., Morris, A. E., Lee, C. C., ... & Lyman, S. D. (1996). Hematologic effects of flt3 ligand in vivo in mice.
- Mueller, S. N., & Germain, R. N. (2009). Stromal cell contributions to the homeostasis and functionality of the immune system. Nature Reviews Immunology, 9(9), 618-629.
- Oh, I. H., & Kwon, K. R. (2010). Concise review: multiple niches for hematopoietic stem cell regulations. Stem cells, 28(7), 1243-1249.
- Fonsatti, E., Sigalotti, L., Arslan, P., Altomonte, M., & Maio, M. (2003). Emerging role of endoglin (CD105) as a marker of angiogenesis with clinical potential in human malignancies. Current Cancer Drug Targets, 3(6), 427-432.
- Newman, P. J. (1997). The biology of PECAM-1. The Journal of clinical investigation, 99(1), 3-8.
- Farr, A. G., Berry, M. L., Kim, A., Nelson, A. J., Welch, M. P., & Aruffo, A. (1992). Characterization and cloning of a novel glycoprotein expressed by stromal cells in T-dependent areas of peripheral lymphoid tissues. The Journal of experimental medicine, 176 (5), 1477-1482.
- Nagasawa, T., Omatsu, Y., & Sugiyama, T. (2011). Control of hematopoietic stem cells by the bone marrow stromal niche: the role of reticular cells. Trends in immunology, 32(7), 315-320.
- Garg, S., Madkaikar, M., & Ghosh, K. (2013). Investigating cell surface markers on normal hematopoietic stem cells in three different niche conditions. International journal of stem cells, 6(2), 129.
- Dominici, M. L. B. K., Le Blanc, K., Mueller, I., Slaper-Cortenbach, I., Marini, F. C., Krause, D. S., ... & Horwitz, E. M. (2006). Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy, 8(4), 315-317.
- Spangrude, G. J., Heimfeld, S., & Weissman, I. L. (1988). Purification and characterization of mouse hematopoietic stem cells. Science, 241 (4861), 58-62.
- Yang, L., Bryder, D., Adolfsson, J., Nygren, J., Månsson, R., Sigvardsson, M., & Jacobsen, S. E. W. (2005). Identification of Lin–Sca1+ kit+ CD34+ Flt3–short-term hematopoietic stem cells capable of rapidly reconstituting and rescuing myeloablated transplant recipients. Blood, 105 (7), 2717-2723.
- Yilmaz, O. H., Kiel, M. J., & Morrison, S. J. (2006). SLAM family markers are conserved among hematopoietic stem cells from old and reconstituted mice and markedly increase their purity. Blood, 107(3), 924-930.
- Mora, J. R., Bono, M. R., Manjunath, N., Weninger, W., Cavanagh, L. L., Rosemblatt, M., & Von Andrian, U. H. (2003). Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature, 424(6944), 88-93.
- Morita, Y., Iseki, A., Okamura, S., Suzuki, S., Nakauchi, H., & Ema, H. (2011). Functional characterization of hematopoietic stem cells in the spleen. Experimental hematology, 39(3), 351-359.
- Ogawa, M., Matsuzaki, Y., Nishikawa, S., Hayashi, S., Kunisada, T., Sudo, T., ... & Nishikawa, S. (1991). Expression and function of c-kit in hemopoietic progenitor cells. The Journal of experimental medicine, 174(1), 63-71.
- Pronk, C. J., Rossi, D. J., Månsson, R., Attema, J. L., Norddahl, G. L., Chan, C. K. F., ... & Bryder, D. (2007). Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell stem cell, 1(4), 428-442.
- Slayton, W. B., Georgelas, A., Pierce, L. J., Elenitoba-Johnson, K. S., Perry, S. S., Marx, M., & Spangrude, G. J. (2002). The spleen is a major site of megakaryopoiesis following transplantation of murine hematopoietic stem cells. Blood, The Journal of the American Society of Hematology, 100(12), 3975-3982.
- Oguro, H., Ding, L., & Morrison, S. J. (2013). SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell stem cell, 13(1), 102-116.
- Pietras, E. M., Reynaud, D., Kang, Y. A., Carlin, D., Calero-Nieto, F. J., Leavitt, A. D., ... & Passegué, E. (2015). Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell stem cell, 17(1), 35-46.
- Purton, L. E. (2022). Adult murine hematopoietic stem cells and progenitors: an update on their identities, functions, and assays. Experimental Hematology, 116, 1-14.
- Yu, H., Gibson, J. A., Pinkus, G. S., & Hornick, J. L. (2007). Podoplanin (D2-40) is a novel marker for follicular dendritic cell tumors. American Journal of Clinical Pathology, 128(5), 776-782.
- Lim, H. K., & O’Neill, H. C. (2019). Identification of stromal cells in spleen which support myelopoiesis. Frontiers in Cell and Developmental Biology, 7, 1.
- Sugiyama, T., Omatsu, Y., & Nagasawa, T. (2019). Niches for hematopoietic stem cells and immune cell progenitors. International immunology, 31(1), 5-11.
- Oda, A., Tezuka, T., Ueno, Y., Hosoda, S., Amemiya, Y., Notsu, C., ... & Goitsuka, R. (2018). Niche-induced extramedullary hematopoiesis in the spleen is regulated by the transcription factor Tlx1. Scientific reports, 8(1), 1-16.
- Periasamy, P., Tran, V., & O’Neill, H. C. (2018). Identification of genes which regulate stroma-dependent in vitro hematopoiesis. PLoS One, 13(10), e0205583.
- Tan, J. K., & O’Neill, H. C. (2010). Haematopoietic stem cells in spleen have distinct differentiative potential for antigen presenting cells. Journal of Cellular and Molecular Medicine, 14(8), 2144-2150.
- Christensen, J. L., Wright, D. E., Wagers, A. J., & Weissman, I. L. (2004). Circulation and chemotaxis of fetal hematopoietic stem cells. PLoS biology, 2(3), e75.
- Petvises, S., & O’Neill, H. C. (2014). Distinct progenitor origin distinguishes a lineage of dendritic-like cells in spleen. Frontiers in immunology, 4, 501.
- Wright, D. E., Cheshier, S. H., Wagers, A. J., Randall, T. D., Christensen, J. L., & Weissman, I. L. (2001). Cyclophosphamide/granulocyte colony-stimulating factor causes selective mobilization of bone marrow hematopoietic stem cells into the blood after M phase of the cell cycle. Blood, The Journal of the American Society of Hematology, 97(8), 2278-2285.
- Morrison, S. J., Wright, D. E., & Weissman, I. L. (1997). Cyclophosphamide/granulocyte colony-stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proceedings of the National Academy of Sciences, 94 (5), 1908-1913.
- Tan, J. K., & Watanabe, T. (2014). Murine spleen tissue regeneration from neonatal spleen capsule requires lymphotoxin priming of stromal cells. The Journal of Immunology, 193(3), 1194-1203.
- Chow, A., Lucas, D., Hidalgo, A., Méndez-Ferrer, S., Hashimoto, D., Scheiermann, C., ... & Frenette, P. S. (2011). Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. Journal of Experimental Medicine, 208(2), 261-271.
- Dutta, P., Hoyer, F. F., Grigoryeva, L. S., Sager, H. B., Leuschner, F., Courties, G., ... & Nahrendorf, M. (2015). Macrophages retain hematopoietic stem cells in the spleen via VCAM-1. Journal of Experimental Medicine, 212(4), 497-512.
- Tang, H., Guo, Z., Zhang, M., Wang, J., Chen, G., & Cao, X. (2006). Endothelial stroma programs hematopoietic stem cells to differentiate into regulatory dendritic cells through IL-10. Blood, 108 (4), 1189-1197.
- Fujisaki, J., Wu, J., Carlson, A. L., Silberstein, L., Putheti, P., Larocca, R., ... & Lin, C. P. (2011). In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature, 474(7350), 216-219.
- Kuper, C. F., van Bilsen, J., Cnossen, H., Houben, G., Garthoff, J., & Wolterbeek, A. (2016). Development of immune organs and functioning in humans and test animals: Implications for immune intervention studies. Reproductive toxicology, 64, 180-190.
- Golub, R., & Cumano, A. (2013). Embryonic hematopoiesis. Blood Cells, Molecules, and Diseases, 51(4), 226-231.
- Steiniger, B., Ulfig, N., Riße, M., & Barth, P. J. (2007). Fetal and early post-natal development of the human spleen: from primordial arterial B cell lobules to a non-segmented organ. Histochemistry and cell biology, 128, 205-215.


