Current Issue : Article / Volume 2, Issue 2
- Case Report | DOI:
- https://doi.org/10.58489/2836-502X/009
Pheochromocytoma Associated with Neurofibromatosis Type 1 Presenting with Secondary Amenorrhea - A Rare Case Report.
1Assistant Professor, 2Professor, 3Associate Professor
1Dept. of General Surgery, A.J.Institute of medical Sciences & Research Centre, Mangalore, India
Anand Bhandary Panambur
AnandB.P., A.Hegde,A.I.Peter,(2022). Pheochromocytoma Associated with Neurofibromatosis Type 1 Presenting with Secondary Amenorrhea - A Rare Case Report. Journal of Endocrine System and Diabetes (JESD). 2(2). DOI: 10.58489/2836-502X/009.
© 2022 Anand Bhandary Panambur, this is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
- Received Date: 24-12-2022
- Accepted Date: 09-01-2023
- Published Date: 19-01-2023
Pheochromocytoma, Neurofibromatosis type-1, secondary amenorrhea, normotensive pheochromocytoma, open adrenalectomy, NF-1
Abstract
Neurofibromatosis type 1 is a complex, multi-system genetic disorder that is associated with an increased prevalence of pheochromocytoma 1.0%â5.7% compared to the general population. Currently, there are no generally accepted guidelines of screening for pheochromocytoma and in asymptomatic individuals of the population with approaches and practices varying considerably between physicians. We are reporting a rare case of normotensive pheochromocytoma associated with neurofibromatosis type 1 presenting with secondary amenorrhea.
Introduction
Pheochromocytoma are chromaffin cell catecholamine secreting tumors originating from the adrenal medulla. They may present in a number of ways from an incidental finding to that of the classical triad of episodic headache, palpitations and diaphoresis due to hypersecretion of catecholamines resulting in episodic hypertension. The incidence of pheochromocytoma among NF1 patients is estimated at 0.1 to 5.7%, with an incidence as high as 20 to 50% among hypertensive NF1 patients [1]. However, secondary amenorrhea, potentially through activation of alpha receptors, has rarely been reported as a presentation [2].
Herein, we report a rare case of normotensive pheochromocytoma associated with neurofibromatosis type 1 presenting with secondary amenorrhea.
Case Report
An 18-year-old female was referred to the outpatient department of General surgery from OBG department with an ultrasound abdomen report suggestive of enlarged right adrenal gland. On enquiry, she gave history of episodic headache for 6 years, vague upper abdominal discomfort for 6 months and amenorrhea for 6 months. Her blood pressure (recorded on multiple occasions) was around 110/70 mmHg with pulse rate 76-84/min in sitting and 106/70 mmHg with pulse rate 82-90/min in standing position. General physical exam revealed multiple well defined hyperpigmented papules present over the chin, bilateral forearm and back with Café-au-lait spots suggestive of Neurofibromatosis 1. Similar lesions were also noted in her grandfather, father and brother. Systemic examinations were non-contributory.
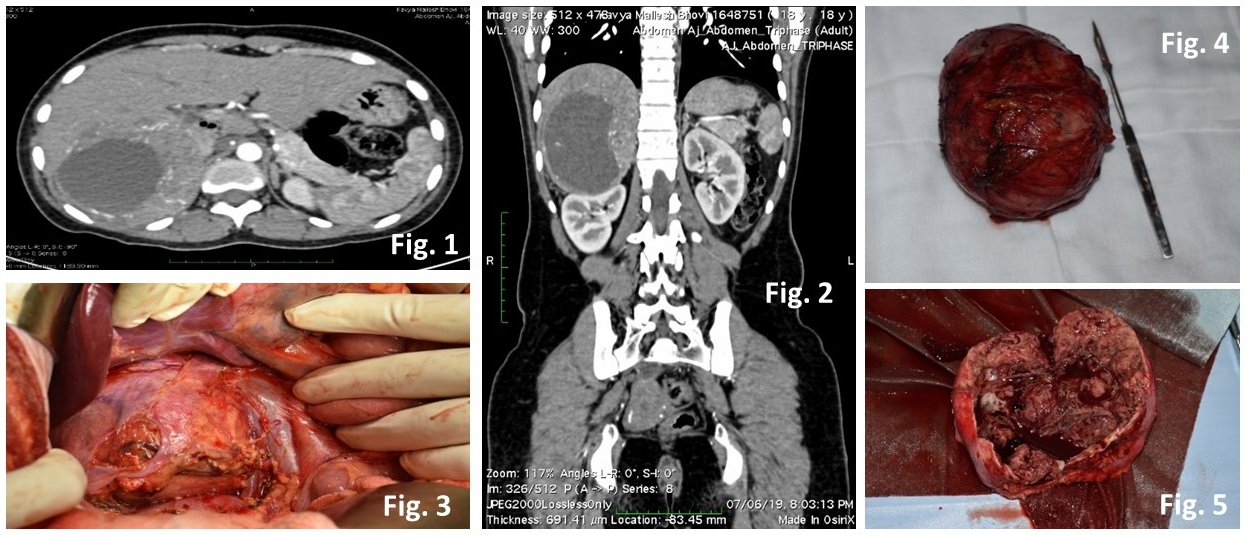
Her routine blood investigations were within normal limits. Subsequently, computed tomography abdomen also confirmed a solid cystic lesion 9.3 x 4.5 x 11.2 cm in right suprarenal location (Fig 1.) displacing the IVC antero-medially with inhomogeneous post contrast enhancement of solid component f/s/o pheochromocytoma (Fig 2.). Her thyroid function tests were within normal limits. Serum cortisol, FSH, LH, 17-OH progesterone and prolactin did not reveal any abnormality. 24-hour urine for VMA was 8.93mg/d (7.9) and 24-hour urine for metanephrine was 2751mcg/d (74-297).
Having thus diagnosed, right adrenal pheochromocytoma with NF-1, the patient was prepared for surgery with volume repletion and administration of extended-release prazosin (2.5 mg) and propranolol (30 mg) in divided doses. Subsequently, she underwent an open right adrenalectomy under general anesthesia (Fig 3.), which she tolerated well. The tumor was well-encapsulated (Fig 4, 5.), and histopathology also confirmed the diagnosis of pheochromocytoma.
Discussion
Neurofibromatosis type 1 is a complex, multi-system genetic disorder that is associated with an increased prevalence of pheochromocytoma 1.0%–5.7% compared to the general population. A delay in pheochromocytoma diagnosis or undiagnosed pheochromocytoma, as seen in normotensive and asymptomatic patients, may portend a significant cardiovascular morbidity and mortality risk due to excess catecholamine secretion [3].
Currently, there are no generally accepted guidelines of screening for pheochromocytoma and in asymptomatic individuals of the population with approaches and practices varying considerably between physicians. Emerging data suggest benefit in routine pheochromocytoma screening of all individuals with neurofibromatosis type 1.
Kepenekian and Gruber suggest biochemical screening in asymptomatic individuals with NF1, with Gruber et al. suggesting screening as routinely as every 3 years starting at age 10–14 years. Every 3-year screening was determined to be sufficient in the population, compared to yearly in VHL and MEN syndromes, as the prevalence is somewhat lower in NF1[4].
Also, rarely pheochromocytoma might present with secondary amenorrhea. One possibility is that ovarian granulsa luteal cells in the human have alpha- and beta-adrenergic receptors. It was shown recently that activation of alpha-adrenergic receptors causes inhibition of LH and HCG mediated progesterone release through intracellular Ca++ and phosphoinositide pathways.
Conclusion
Pheochromocytoma presenting with hypotension and atypical presentation of amenorrhea is a rare condition by itself. The rare possibility of syndromic association should not be missed when patients present to us with such conditions as syndromes associated with genetic disorders may have varied presentation. Genetic syndromes have to be evaluated by gene mapping for a confirmed diagnosis and treatment.
Disclosures
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Funding Sources
The authors did not receive any funding.
References
- Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM (1999). von Recklinghausen’s disease and pheochromocytomas.J Urol;162:1582-1586.
- Fohr KJ, Mayerhofer A, Sterzik K, Rudolf M, Rosenbusch B, Gratzl M (993): Concerted action of human chorionic gonadotropin and norepinephrine on intracellular-free calcium in human granulosa- lutein cells: evidence for the presence of a functional alpha-adrenergic receptor. J Clin Endocrinol Metab 1, 76(2):367-373.
- Ross EJ, Prichard BN, Kaufman L, Robertson AI, Harries BJ (1967). Preoperative and operative management of patients with phaeochromocytoma. Br Med J;1:191-8.
- Gruber LM, Erickson D, Babovic-Vuksanovic D, Thompson GB, Young WF Jr, Bancos I (2017). Pheochromocytoma and paraganglioma in patients with neurofibromatosis type 1. Clin Endocrinol.;86(1):141–9.
- Ammar Wakil* and Stephen Atkin (2008). Secondary amenorrhoea due to pheochromocytoma: a case report Cases Journal, 1:30


