Article In Press : Article / Volume 3, Issue 1
- Research Article | DOI:
- https://doi.org/10.58489/2836-5003/011
Omicron B.A 2: Vaccine Construction with MHC Class-I, MHC Class-II by Using Bioinformatics Tools
1Department of Medicine and Surgery, Al Nafees Medical College Islamabad, Pakistan.
2Department of Computer Science, University of Agriculture Faisalabad, Pakistan.
3Department of Medicine and Surgery, CMH Multan Institute of Medical Sciences, Pakistan.
4Department of Pharmacology and Therapeutics, HITEC, Institute of Medical Sciences Taxila Cantt, Pakistan.
5Department of Medicine and Surgery, Sargodha Medical College, Pakistan.
6Department of Medicine and Surgery, HITEC, Institute of Medical Sciences Taxila Cantt, Pakistan.
7Department of Bioinformatics and Biotechnology, Government College University, Faisalabad, Pakistan.
Muhammad Waqar Mazhar*
Muhammad Waqar Mazhar, et.al., (2024). Omicron B.A 2: Vaccine Construction with MHC Class-I, MHC Class-II by Using Bioinformatics Tools. Archives of Immunology Research and Therapy, 3(1). DOI: 10.58489/2836-5003/011
© 2024 Muhammad Waqar Mazhar, this is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
- Received Date: 28-04-2024
- Accepted Date: 01-05-2024
- Published Date: 06-05-2024
SARS (COV-2), omicron, BA.2, MHC class-I, MHC class-II vaccine
Abstract
SARS (COV-2) was recognized in 31/December/2019 in Wuhan city in the Hubei Provence of China. The omicron variant of COVID-19 reported in November 2021. Omicron can have many earlier documented mutations of other variant of concern (VOC) omicron is including 32 mutations of spike like protein, it is too compared of the 16 mutations. The omicron variant has been divided into four linages BA.1, BA.2, BA.3, B.1.1.529. BA.1 which could destroyed our immune system and vaccine are not highly effective against BA.2.
Introduction
The outbreak of the novel SARS (COV-2) was recognized in 31/December/2019 in Wuhan city in the Hubei Provence of China (MAZHAR, MAHMOOD, SAIF, SHAHZADI, & ASLAM, 2021). The name of corona derived from their crown like spike protein which is attach to the cell receptor. This virus belongs to the coronaviradae family and nideovirales Oder (Mazhar et al., 2022). These cases were increase day by day in Wuhan and Hubei Provence. WHO is declared this epidemic as in the Public Health Emergencies. When the disease is appeared is always a complex situation. Imported and the secondary cases has been reported more than 26. The identification of new emergent omicron (BA.1.1.529) The SARS-COV-2 variant was first introduced in South Africa. The omicron variant of COVID-19 reported in November 2021.The variant was rapidly spread and 72% was shortly thereafter and introduced in infected individuals in South Africa (Andeweg et al., 2022). The spreading of omicron increase rapidly must be increase fast transmissibility of virus. Omicron can have many earlier documented mutations of other variant of concern (VOC) omicron is including 32 mutations of spike like protein, it is too compared of the 16 mutations (He, Hong, Pan, Lu, & Wei, 2021) which is highly infectious delta version, as for as other proteins are needed for the viral replication like NSP12 and NSP14. Spike mutations which can have increase the spike ability to connect ACE2 receptor on the host cells (Yoshimoto, 2021).
The omicron variant has been divided into four linages BA.1, BA.2, BA.3, B.1.1.529. BA.1 which could destroyed our immune system and vaccine are not highly effective against BA.2 (Chen & Wei, 2022). It is mild version the severity and death were less. BA.2 sequences in GISAID were also reported in Denmark. A total 89 sequences reported of BA.2 has been uploaded in GISAID (Hirotsu et al., 2022). In this study BA.1 has 39 mutations. BA.2 has 31 mutations.BA.3 has 34 mutations.BA.1.1 has 40 mutations (Kumar, Karuppanan, & Subramaniam, 2022).
BA.2 is more pathogenic than BA.1, BA.2 sequences which was uploaded from south Africa on November 2021.The case was first study and discovered in China (Ferenčak et al., 2022). The transmissibility and biological characteristics were quite different because of their different genome.BA.2 is founded to be able to alarming to reinfect patient which is infected by omicron BA.2(Chen & Wei, 2022). As we found that omicron sub-variants have a higher positive electrostatic surface potential. An important question is that BA.2 or BA.3 will be the new dominating concern variant (Mueller). The WHO has been declared the outbreak caused by SARS- COV-2 as a global pandemic. As in November 2021, more than 258,483,028 infections and 6,135,013 as worldwide deaths had been reported (Awadasseid, Wu, Tanaka, & Zhang, 2021).
Methodology
The whole phenomena for designing vaccine of BA.2 (omicron) a variant of severe acute respiratory syndrome coronavirus2 (SARC-CoV2) is based on five major steps which are (1) sequence retrieval and its structure analysis (2) Epitopsis prediction (B&T-cell epitopsis prediction) (3) Vaccine Construction (4) Secondary & Tertiary structure Extrapolation and Validation (5) Molecular Dynamics & Expression Analysis. For constructing potential vaccine, nucleocapsid phosphoprotein sequence of omicron having Accession no. UJP23613.1 is retrieved from NCBI (https://www.ncbi.nlm.nih.gov/), the sequence then put into ExPAsy Protpam tool to figure out the physiochemical properties of target protein. Secondary structure of protein predicted via PSIPRED Tool (https://www.expasy.org/). The antigen probability of protein was checked by vaxijen V2.0. For higher specificity, the threshold value was set at ≥ 0.4. the allergenicity of protein was checked by AllerTOP V2.0. The tertiary structure of Omicron protein was checked by using trRoasetta (https://yanglab.nankai.edu.cn/trRosetta/). The epitopes were predicted by using Bepipred Linear Epitope Prediction. The tool uses Hidden Markov model-based algorithm, a one of the best linear epitopes calculating method. Homo sapiens were selected as MHC source the ANN 4.0 method to investigate diverse set of MHC HLA alleles in humans. IEBD tool supplies an output interface for HLA-binding affinity of epitopes according to IC50 nM unit.For constructing complete set of vaccine 5 B-cell epitopes, 12 epitopes of T-cell for MHC-Class I and 19 epitopes for MHC-Class II respectively were used. the antigenicity of target protein with and without adjuvant was calculated via VaxijenV2.0. The vaccine was founded to be non-allergenic via using AllerTopV2.0 tool. Exploration of secondary and tertiary structure of our construct was checked via PRISPRED Tool and TrRosseta respectively. Refinement of 3D structure of our construct was checked via GalaxyRefine tool. The refinement of 3D structure validates through UCLA-DOE-LAB server. Molecular docking of refined vaccine model using toll-like receptor 3 ligand as a binding domain having accession number 2A0Z on Protein data Bank (Pdb) was performed via using Cluspro V 2.0 for protein-protein docking.
Results
sequence retrieval and its structure analysis
The result prove that total number of amino acids is 416, molecular weight (45409.59), theoretical isoelectric point pI(10.13), Total number of negatively charged residues (Asp + Glu): 35 Total number of positively charged residues (Arg + Lys): 61, Total number of atoms: 6332. The chemical formula which is gained C1965H3133N605O622S7 Instability index (II) is computed to be 55.74 which classify the target protein as unstable, Aliphatic index 53.85 and Grand average of hydropathicity (GRAVY): -0.962. The result (as shown in fig.1) clearly says that alpha helix, beta strands, coils and extracellular region are present in protein sequence and mostly nonpolar in nature having a good confident score. There was no disulfide bonds present between the amino acids. The Overall Prediction for the Protective Antigen value was 0.5038, which encourage us that this protein was able to make antigen.
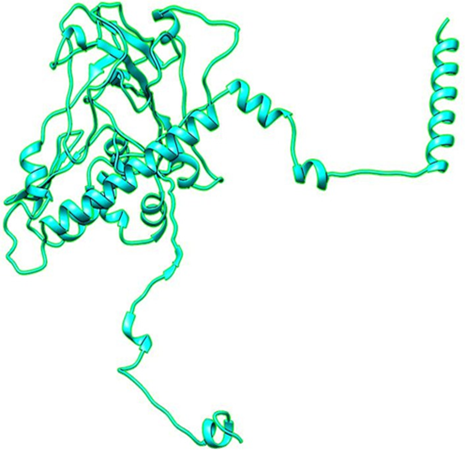
Fig1: 3D structure of nucleocapsid phosphoprotein
The results shows that the protein was non allergenic. The results shown in fig.1.The predicted model is medium with TM-score 0.439. The predicted sequence was 42.8% disordered but reliability for disordered region is low in general.
Epitopsis prediction
An epitope, also called antigenic determinant, is a part of an antigen which is found by using the immune system, with the aid of using antibodies like as B cells, or T cells. The epitope is the precise piece of the antigen to whom an antibody bind. A part of an antibody that binds to the epitope is known as a paratope. Although epitopes are normally non-self-proteins, sequences derived from the host that may be found also are epitopes (12). Here, we use immune epitope data base (IEDB) methods for the analysis of potential B-cell epitopes and T-cell epitopes, respectively.
B-cell epitopes prediction
The smallest score for linear epitope prediction was 0.296 and the largest score was 0.744. Moreover, the average prediction score was found to be 0. 558.Out of 9, Total 5 epitopes were found to be satisfying the antigenicity and non-allergenic analysis. Table for whole epitopes was given in S1 table and the candidate peptides are given in table 1. Moreover, these 5 peptides were found to have potential to start B-cell response. Total 5 selected epitopes were used in conservancy analysis that were choose to be utilized in vaccine construction.
Table 1: List of Bepipred Linear Epitope predicted using IEDB analysis resource.
| Start | End | Peptide | Length | Antigenicity | Allergenicity |
| 4 | 45 | NGPQNQRNALRIT FGGPSDSTGSNQN GGARSKQRRPQG LPNN | 42 | 0.4236 | NON-ALLERGEN |
| 56 | 102 | HGKEDLKFPRGQ GVPINTNSSPDDQI GYYRRATRRIRGG DGKMKDLS | 47 | 0.5773 | NON-ALLERGEN |
| 224 | 264 | LNQLESKMSGKG QQQQGQTVTKKS AAEASKKPRQKRT ATKA | 41 | 0.5529 | NON-ALLERGEN |
| 273 | 296 | RRGPEQTQGNFG DQELIRQGTDYK | 24 | 0.6277 | NON-ALLERGEN |
| 340 | 345 | DPNFKD | 6 | 2.8780 | NON-ALLERGEN |
Lymphocytes present on their surface a particular receptor known as T-cell receptor (TCR) that empowers the acknowledgment of antigens when they are shown on the outer layer of antigen- introducing cells (APCs) bound to significant histocompatibility complex (MHC) atoms. T-cell epitopes are introduced by class I (MHC I) and II (MHC II).
MHC Class-I
A lower IC50 value figures out higher binding affinity of epitopes with MHC Class-I molecule. Total 126 number of epitopes were selected based on IC50 values less than 100 for certifying higher affinity estimated to interact with many MHC Class-1. Out of 126 epitopes, 40 were selected based on their probability of making antigen and non-allergenicity and then some of them also screened out on the behalf of their toxicity. 12 epitopes of MHC-Class 1 were selected for further analysis. The epitopes KTFPPTEPK, AQFAPSASA, LSPRWYFYYL, SPRWYFYYL were predicted to be dominant binder with alleles (HLA-A*11:01, HLA-A*30:01, HLA-A*03:01, HLA-A*31:01, HLA-A*68:01, HLA-A*02:06, HLA-B*15:01, HLA-B*08:01, HLA-B*07:02,
HLA-B*07:02, HLA-B*08:01). Moreover, to find out worldwide coverage for MHC-Class1 allele epitopes, population coverage analysis was performed. The computed results prove the 89.98% coverage for entire world, 69.02% for South Africa, 95.83% for Europe and 78.77% for South Asia, respectively.
MHC Class- II
600 conserved predicted epitopes with the IC50 less than 100 were found to interact with MHC Class-II alleles. Out of 600 epitopes, 38 were selected that were interacting with more than 6 MHC Cass-II alleles. 19 epitopes were finalized for further analyses because of their allergenicity, toxicity and antigenicity. The core epitopes AALALLLLDRLNQLE is the top binder as it interacts with 18 alleles; (HLA-DRB4*01:01, HLA-DPA1*03:01/DPB1*04:02, HLA- DRB3*01:01, HLA-DRB1*12:01, HLA-DPA1*02:01/DPB1*01:01, HLA-DRB1*13:02, HLA- DRB1*11:01, HLA-DPA1*02:01/DPB1*05:01, HLA-DRB1*04:01, HLA-DQA1*01:01/DQB1*05:01, HLA-DRB1*03:01, HLA-DPA1*01:03/DPB1*04:01, HLA-
DRB1*04:05, HLA-DRB1*08:02) respectively. Moreover, to check out worldwide coverage for MHC-Class- II allele epitopes, population coverage analysis was performed. The computed results prove the 72.62% coverage for entire world, 32.1% for South Africa, 76.69% for Europe, and 49.66% for South Asia,51.6% for Northeast Asia,76.07% for East Asia, respectively.
Vaccine Construction
50S ribosomal protein L7/L12 with UniProt having ID: P9WHE3 (Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv)) was used as an adjuvant to build the vaccine construct. The adjuvant was linked to the first B-cell epitope by using a linker EAAAK. Furthermore, to link B- cell epitopes with MHC-Class I epitopes, a linker GPGPG was utilized. MHC Class-II epitopes were connected using AAY linkers. For identification and purification process of protein, a 6x His Tag was used at the C-terminus of vaccine sequence as shown in Fig2. The final sequence for vaccine construct with the molecular weight 77904.42 and 708 amino acids. The complete vaccine sequence was given in supplement paper. the antigenicity was estimated 0.6327 without using adjuvant and 0.5833 by using 50S ribosomal adjuvant in protein sequence. It was concluded from the results that the vaccine construct is antigenic in nature either it is attached with adjuvant or not.
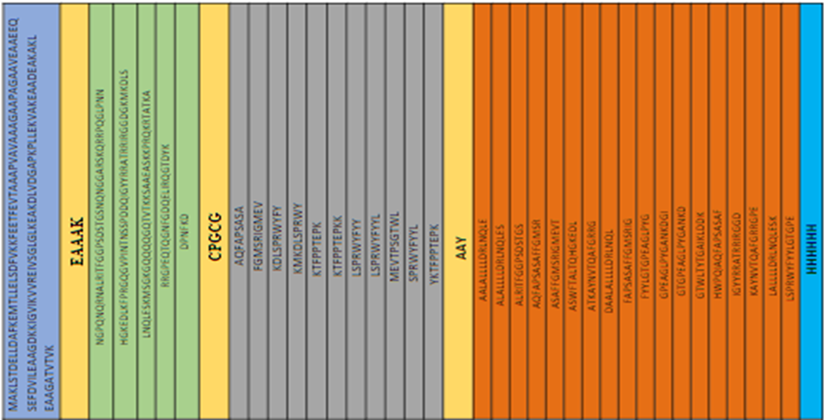
Fig 2: Blue color shows 50S ribosomal protein L7/L12, Skin color shows linkers, green color shows B-cell epitopes, grey color shows MHC-Class 1, orange color shows MHC-Class2 and sky-blue color shows 6x His tag.
The physiochemical properties of target vaccine sequence were checked via ExPasy Protpam tool. The Molecular weight of multi-epitope subunits were 77904.42. Calculated PI of protein was 9.46. The instability index (II) is computed to be 48.05. hydropathicity (GRAVY) was -0.489. Our vaccine construct shows high solubility rate computed through Soluprot with the estimated score 0.587.
Secondary & Tertiary structure Extrapolation
Most of the region is coiled, it took almost 95% of the structure, while 3%of the structure contains alpha-helix and 2%is beta-sheets. Following figures Clarify the positions of amino acids.
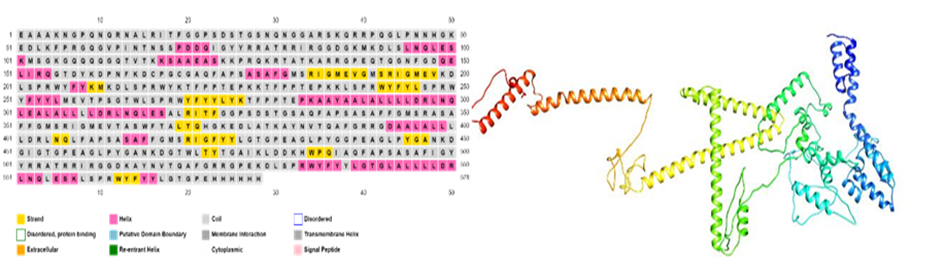
Fig 3(a): shows secondary structure of constructed vaccine Fig 3(b):Tertiary structure
Validation of Refined 3D structure
five models of the vaccine chimera after refinement. Various parameters were considered in the process of refinement such as GDT-HA (0.9784), RMSD (0.341), and MolProbity (1.290). The calculation of clash score was found to be 5.4, score of poor rotamers was 0.6 and the Ramachandran score was predicted as 99.0%. Model 1 was selected according to above parameters as it was most suitable among other 4. 3D structure of refined vaccine added in supplement.
The structure was analyzed and Ramachander plot was produced. There was total 96.4% region, that was in favored region of the plot represented by red color in given figure whereas, 3% structure was plotted in additional allowed region (yellow color),0% in general allowed region,0.6% in disallowed region.
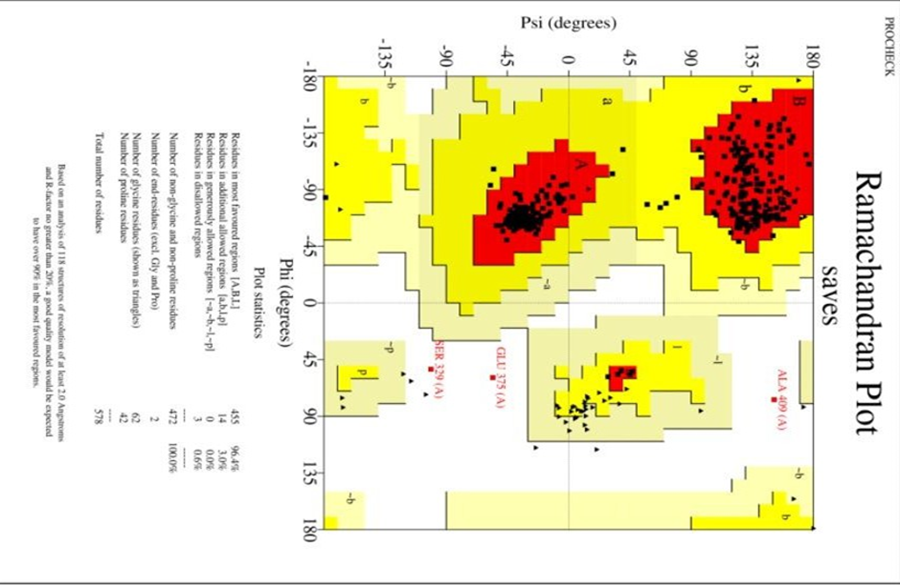
Fig 4.
Molecular Dynamics & Expression Analysis
Out of 30 docked models, model no.0 proved to be best docked model as this model had -902.4 molecular weighted score. The Figure 5 shows the molecular docking complex.
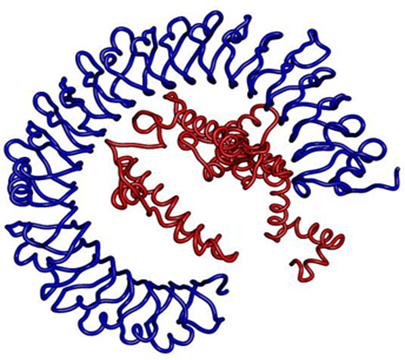
Fig 5: Docked complex of vaccine with toll-like receptor 3 (TLR3)
The optimized codon had a length of 1738 nucleotides and it displayed that codon adaptation index (CAI) for optimized sequence was 0.64 with GC content of 67.88%. These values were expressed in stable vector E. coli (strain 12K), as optimize GC content ranges between 30 to 70%. Finally, the optimized sequence was cloned in pET28 (+) vector to make a recombinant plasmid after being amplified through in-silico PCR by using SnapGene software.
Discussion
This study proves that BA.2 have total 416 amino acids, molecular weight (45409.59), theoretical isoelectric point pI(10.13), total number of negatively charged residues (Asp + Glu): 35 (Kaukinen, Koistinen, Vapalahti, Vaheri, & Plyusnin, 2001), total number of positively charged residues (Arg + Lys): 61, total number of atoms: 6332. The chemical formula which is gained C1965H3133N605O622S7. Instability index (II) is computed to be 55.74 which classify the target protein as unstable.
The results shows that in MHC class-I epitopes KTFPPTEPK, AQFAPSASA, LSPRWYFYYL, SPRWYFYYL were predicted to be dominant binder with alleles (HLA-A*11:01, HLA-A*30:01, HLA-A*03:01, HLA-A*31:01, HLA-A*68:01, HLA-A*02:06, HLA-B*15:01, HLA-B*08:01,
HLA-B*07:02, HLA-B*07:02, HLA-B*08:01). toxicity and antigenicity. in MHC class-II core epitopes AALALLLLDRLNQLE is the top binder as it interacts with 18 alleles; (HLA- DRB4*01:01, HLA-DPA1*03:01/DPB1*04:02, HLA-DRB3*01:01, HLA-DRB1*12:01, HLA- DPA1*02:01/DPB1*01:01, HLA-DRB1*13:02, HLA-DRB1*11:01, HLA-DPA1*02:01/DPB1*05:01, HLA-DRB1*04:01, HLA-DQA1*01:01/DQB1*05:01, HLA- DRB1*03:01, HLA-DPA1*01:03/DPB1*04:01, HLA-DRB1*04:05, HLA-DRB1*08:02)
respectively. The Molecular weight of multi-epitope subunits were 77904.42. Calculated PI of protein was 9.46. The instability index (II) is computed to be 48.05. hydropathicity (GRAVY) was
-0.489. Our vaccine construct shows high solubility rate computed through Soluprot with the estimated score 0.587.
References
- Andeweg, S. P., de Gier, B., Eggink, D., van den Ende, C., van Maarseveen, N., Ali, L., ... & Knol, M. J. (2022). Protection of COVID-19 vaccination and previous infection against Omicron BA. 1, BA. 2 and Delta SARS-CoV-2 infections. Nature Communications, 13(1), 4738.
- Awadasseid, A., Wu, Y., Tanaka, Y., & Zhang, W. (2021). Current advances in the development of SARS-CoV-2 vaccines. International journal of biological sciences, 17(1), 8.
- Chen, J., & Wei, G. W. (2022). Omicron BA. 2 (B. 1.1. 529.2): high potential for becoming the next dominant variant. The journal of physical chemistry letters, 13(17), 3840-3849.
- Ferenčak, I., Obrovac, M., Žmak, L., Kuzle, J., Petrović, G., Vilibić-Čavlek, T., ... & Tabain, I. (2022). SARS-CoV-2 Omicron Variant in Croatia—Rapid Detection of the First Case and Cross-Border Spread. Pathogens, 11(5), 511.
- He, X., Hong, W., Pan, X., Lu, G., & Wei, X. (2021). SARS‐CoV‐2 Omicron variant: characteristics and prevention. MedComm, 2(4), 838-845.
- Hirotsu, Y., Maejima, M., Shibusawa, M., Natori, Y., Nagakubo, Y., Hosaka, K., ... & Omata, M. (2022). Classification of Omicron BA. 1, BA. 1.1, and BA. 2 sublineages by TaqMan assay consistent with whole genome analysis data. International Journal of Infectious Diseases, 122, 486-491.
- Kaukinen, P., Koistinen, V., Vapalahti, O., Vaheri, A., & Plyusnin, A. (2001). Interaction between molecules of hantavirus nucleocapsid protein. Journal of General Virology, 82(8), 1845-1853.
- Kumar, S., Karuppanan, K., & Subramaniam, G. (2022). Omicron (BA. 1) and sub‐variants (BA. 1.1, BA. 2, and BA. 3) of SARS‐CoV‐2 spike infectivity and pathogenicity: A comparative sequence and structural‐based computational assessment. Journal of medical virology, 94(10), 4780-4791.
- MAZHAR, M. W., MAHMOOD, J., SAIF, S., SHAHZADI, M. B., & ASLAM, H. (2021). Severe acute respiratory syndrome (Saras Covid-2): a comprehensive review. Quantum Journal of Medical and Health Sciences, 1(4), 25-33.
- Mazhar, M. W., Raza, A., Zubair, M., Mahmood, J., Tahir, H., Saif, S., & Mazhar, F. (2022). Detection of SARS-CoV-2 by using real-time PCR nasopharyngeal swabs in suspected patients and their clinical medication. Sensors International, 3, 100148.
- Mueller, M. COVID-19 Evidence Support Team EVIDENCE SEARCH REPORT. Scientific American, 2022, 04- 04.
- Yoshimoto, F. K. (2021). A biochemical perspective of the nonstructural proteins (NSPs) and the spike protein of SARS CoV-2. The protein journal, 40(3), 260-295.


