Current Issue : Article / Volume 3, Issue 1
- Case Report | DOI:
- https://doi.org/10.58489/2836-8851/015
Granulomatosis with Polyangiitis (Wegenerâs Granulomatosis) Manifesting with Cranial Nerve Palsies and Pachymeningitis: A Case Report.
- Hôpital La Timone, France.
Christian Matta
Christian Matta. (2024). Granulomatosis with Polyangiitis (Wegenerâs Granulomatosis) Manifesting with Cranial Nerve Palsies and Pachymeningitis: A Case Report. Neurons and Neurological Disorders. 3(1); DOI: 10.58489/2836-8851/015
© 2024 Christian Matta, this is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
- Received Date: 23-12-2023
- Accepted Date: 01-03-2024
- Published Date: 13-03-2024
Abstract
We present a case of a 78-year-old male patient diagnosed with pachymeningitis secondary to Wegener's granulomatosis (WG), also known as granulomatosis with polyangiitis (GPA), associated with p-ANCA positivity. This case report aims to highlight the clinical presentation and diagnosis, of pachymeningitis in WG and provide a review of the literature on this manifestation.
Introduction
Wegener's granulomatosis (WG) is a systemic small-vessel vasculitis characterized by necrotizing granulomatous inflammation, primarily affecting the upper and lower respiratory tracts and kidneys [1].
Central nervous system (CNS) involvement is uncommon, occurring in approximately 2-8% of cases, with the most common manifestation being peripheral neuropathy. However, pachymeningitis is a rare manifestation of GPA, characterized by thickening of the dura mater [2,3].
We present a case of pachymeningitis due to GPA, providing simultaneously a literature review.
Case Presentation
In February 2020, a 78-year-old male patient presented with binocular diplopia, bitemporal headache and a progressive vision loss in the left eye accompanied by a bilateral serous otitis media. A brain computed tomography (CT) was performed, however no significant findings were found.
After a progressive degradation of his visual field, a brain MRI with gadolinium enhancement was performed in 2021 and demonstrated a subdural hematoma of the left convexity with a diffuse, thick dural enhancement suggestive of pachymeningitis (Figure 1).
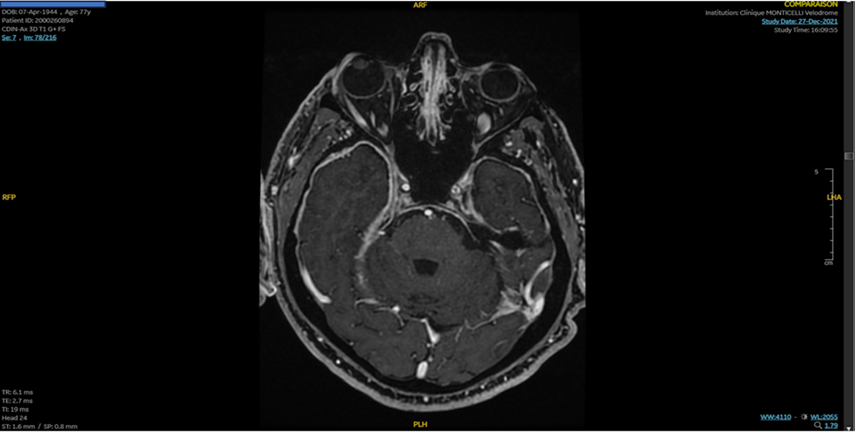
A set of laboratory tests was performed to including autoimmune antibodies: anti-nuclear antibodies, antibodies against extractable nuclear antigens, anti-double- and single-stranded DNA; anti-neutrophil cytoplasmic antibodies, anti-cyclic citrullinated peptide antibodies, anti-MOG, and anti-aquaporin-4 (AQP4), all proved negative.
The blood levels of vitamin B12, B9, thyroid-stimulating hormone, and the angiotensin-converting enzyme (ACE) were normal.
Lumbar puncture revealed an opening pressure of 20 cmH2o, elevated protein of 1.3g/L, normal glucose levels and normal WCC in the cerebrospinal fluid (CSF), with a sterile culture.
In March 2022, the patient presents a progressive onset of visual loss in the right eye. Ophthalmic examination showed a visual acuity at 2/10 without papillitis. A brain MRI found pachymeningitis with atrophy of the left optic nerve (Figure 2).
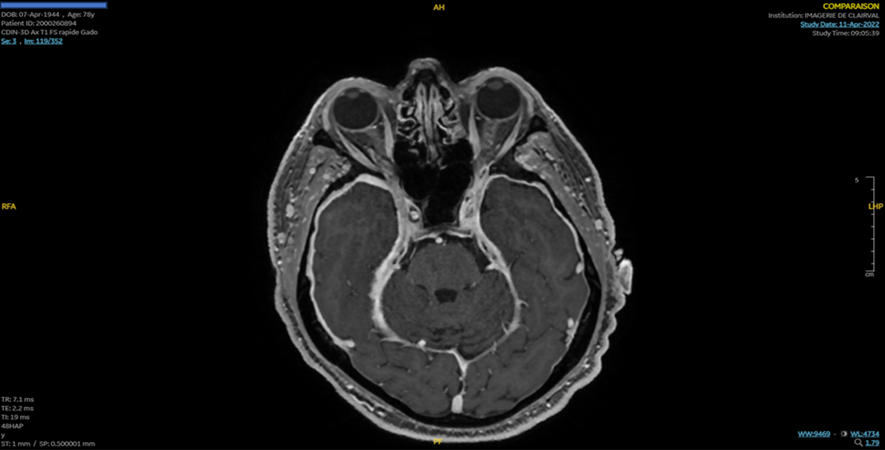
A CT scan of the chest showed the presence of a nodular shadow with some cavitation involving the middle lobe of the left lung in favor of vascular malformation. Salivary gland biopsy showed sialadenitis. A blood patch for suspicion of CSF hypotension was performed as a therapeutic test but without clinical benefit.
In May 2022, a brain and whole-body FDG-PET was performed and showed the presence of moderately hypermetabolic bilateral hilar lymph nodes, and the progression of left frontal pachymeningitis associated with moderate hypermetabolism affecting bilateral internal temporal regions.
The patient was started on intravenous methylprednisolone (1 g/day) for 3 days, without ocular improvement and with poor tolerance of his diabetes
The patient was re-hospitalized for a follow-up evaluation in January 2023.
A whole-body Positron emission tomography (PET) imaging with F-18-labelled 2-fluoro 2-deoxy-D glucose (F-18 FDG) showed a hypermetabolic nodule of the walls of the trachea, the right main bronchus, the subcarinal, the pulmonary hiles, and the nasal septum. A cerebral F-18 FDG PET showed intense hypermetabolism of the right optic nerve, associated with marked encephalic hypermetabolism, predominantly in the temporal and supratentorial regions (Figure 3 and 4).
Laboratory test showed Erythrocyte sedimentation rate (ESR) of 65 mm/h (normal range: 0-20 mm/h), C-reactive protein (CRP) of 45 mg/L (normal range: <10>
Autoimmune and infectious tests were re-performed, and all proved negative including the Anti-neutrophil cytoplasmic antibodies directed to proteinase3 (c-ANCA) but not the Perinuclear anti-neutrophil cytoplasmic antibodies (P-ANCA) which was positive.
So, the patient was diagnosed with granulomatosis with angiitis.
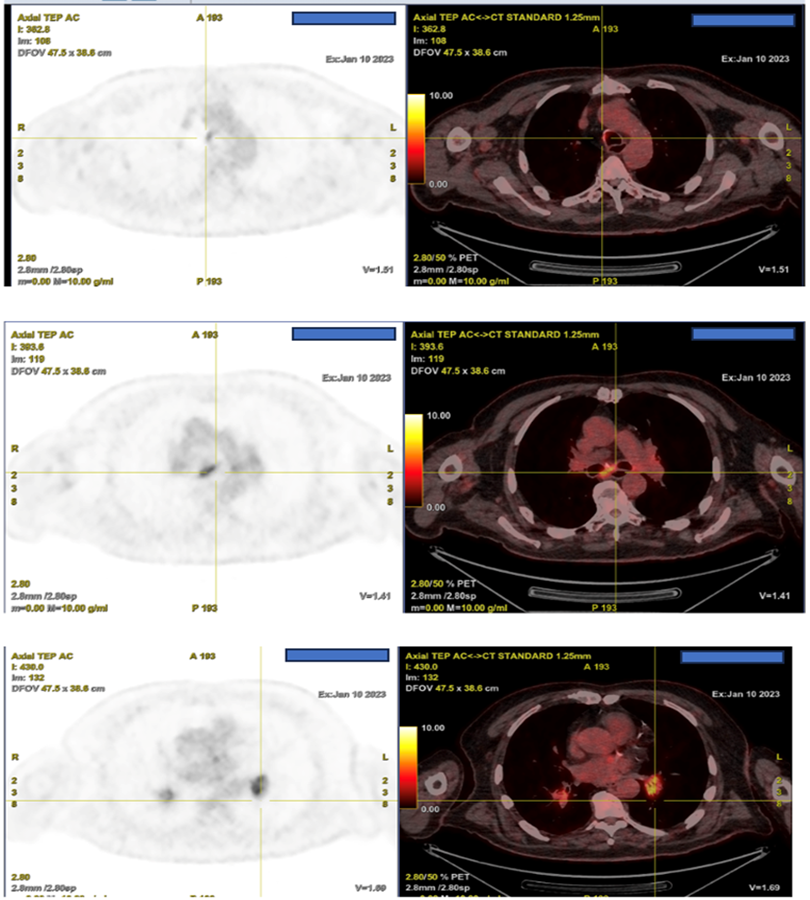
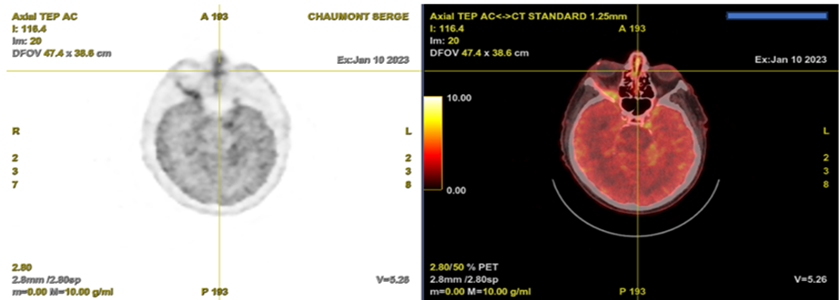
Discussion
Previously known as Wegener’s Granulomatosis, this systemic necrotising vasculitis is associated with cANCA PR3. Predominantly afflicting small vessels, the multisystem disease is classically recognized to affect the renal system alongside upper and lower respiratory tracts [4].
Pachymeningitis, a rare manifestation of granulomatosis with polyangiitis (GPA). It can present with various neurological symptoms and is associated with ANCA (antineutrophil cytoplasmic antibody) positivity. It was reported by Nagashima et al. (2000). Where they presented a case study of a patient with P ANCA Wegeners granulomatosis who had pachymeningitis and multiple cranial neuropathies [5].
This case report discussed the association of pachymeningitis with P-ANCA-positive Wegener's granulomatosis, associated with the imaging pattern of WG on 18-FDG PET Scan. It emphasizes the diagnostic challenge and the need for timely management. These findings underscore the significance of recognizing pachymeningitis in the context of GPA and its association with p ANCA positivity for appropriate diagnosis and management. It highlights the importance of retesting for ANCA when symptoms and imaging are highly suggestive of the diagnosis.
In conclusion, pachymeningitis is a rare and challenging manifestation of WG. A high index of suspicion, along with a combination of clinical, laboratory, and radiological findings, is essential for accurate diagnosis and prompt initiation of appropriate treatment.
References
- Schilder, A. M. (2010). Wegener's granulomatosis vasculitis and granuloma. Autoimmunity reviews, 9(7), 483-487.
- Burrell, H. C., & McConachie, N. S. (1998). Pachymeningitis in Wegener's granulomatosis. Australasian radiology, 42(4), 364-366.
- Sakellariou, G. T., & Kefala, N. (2013). Pachymeningitis in granulomatosis with polyangiitis: a case report and a review of the literature. Case Reports in Rheumatology, 2013.
- Trickey, J., Abbas, A., Lawley, A., Dixey, J., & Jumma, O. (2016). Hypertrophic cranial pachymeningitis and granulomatosis with polyangiitis. QJM: An International Journal of Medicine, 109(4), 267-268.
- Nagashima, T., Maguchi, S., Terayama, Y., Horimoto, M., Nemoto, M., Nunomura, M., ... & Nagashima, K. (2000). P‐ANCA‐positive Wegener’s granulomatosis presenting with hypertrophic pachymeningitis and multiple cranial neuropathies: Case report and review of literature. Neuropathology, 20(1), 23-30.


